Modeling the effects of EMT-immune dynamics
Behind the paper
Modeling the effects of EMT-immune dynamics on carcinoma disease progression
Daniel R. Bergman, Matthew K. Karikomi, Min Yu, Qing Nie & Adam L. MacLean
Read the paper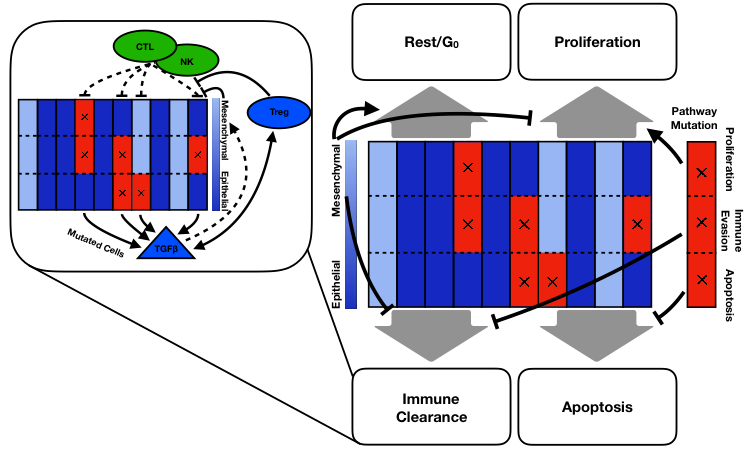
Figure 1: Model Cartoon. Each column represents a tumor cell and each row corresponds to a possible pathway mutation. The shade of blue indicates whether the cell is labeled epithelial or mesenchymal. Red with an x indicates a pathway mutation. Inset shows effects of immune compartment on tumor cells. Dashed arrows indicate stochastic effects.
Interacting with the tumor cells are immune cells consisting of three cell types: natural killer cells (NKs), cytotoxic T cells (CTLs), and T regulatory cells (Tregs). As we did not consider the role of heterogeneity within each of these cell types, these are modeled as continuous variables, i.e. not agents. CTLs and Tregs are recruited to the TME as tumor cells die. NKs and CTLs clear tumor cells while Tregs stymie them and also release TGFB. Seeking to understand how the mesenchymal phenotype affects progression, we saw that an intermediate level of mesenchymal proliferation maximized the time to progression, i.e. was a best case scenario for the patient. Now, high levels of mesenchymal proliferation would clearly be bad for our in silico patients as then they are basically epithelial cells with immune evasiveness. So what was surprising is that it is worse for the patient to have non-proliferative mesenchymal cells rather than slow-cycling mesenchymal cells.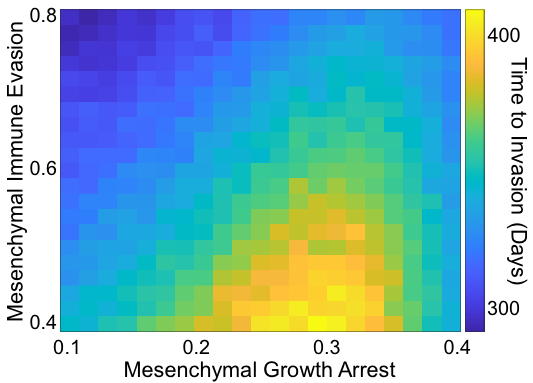
Figure 2: Summary of the effects of mesenchymal parameters on invasion-free survival.
To test this prediction in the model, we employed a Bayesian data analysis framework using gene expression data from TCGA. We found that mesenchymal-associated genes predicted differences between high and low disease-free intervals (DFIs). Many of these genes have known effects on tumor progression, such as both canonical and non-canonical Wnt signaling5. We also discovered new interactions between fibroblast growth factor (FGF) signaling and other transcription factors, the co-regulation of which could affect clinical outcomes, which could be exploited by novel therapeutic strategies.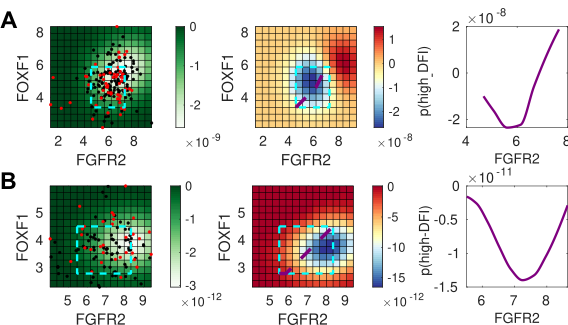
Figure 3:Bayesian analysis predicts the impact of co-expressed gene pairs. For (A) bladder cancer (BLCA), and (B) uterine cancer (UCEC), left panel: the co-expression of FGFR2 and FOXF1; the cyan box indicates the inferred region of high density where predictions can be made. Middle panel: the posterior log probability of high DFI (the desirable outcome), showing the relationship between gene co-expression and positive clinical outcomes. Right panel: 1D slice through the co-expression plot, corresponding to purple line shown on middle panel. The Bayesian analysis predicts that outcomes for BLCA (but not for UCEC) will improve overall with the co-expression of the FGFR2-FOXF1 gene pair.
Within this hybrid multiscale evolutionary ABM framework, we are able predict new mechanisms by which these tumors can be targeted. We also qualified how tumor heterogeneity benefits the tumor, showing yet again that the complexities of tumor biology are necessary to consider in order to optimize treatment.References
- Schreiber, R.D., Old, L.J. and Smyth, M.J., 2011. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science, 331(6024), pp.1565-1570.
- Lim, J. and Thiery, J.P., 2012. Epithelial-mesenchymal transitions: insights from development. Development, 139(19), pp.3471-3486.
- Nieto, M.A., Huang, R.Y.J., Jackson, R.A. and Thiery, J.P., 2016. EMT: 2016. Cell, 166(1), pp.21-45.
- Woods, K., Pasam, A., Jayachandran, A., Andrews, M.C. and Cebon, J., 2014. Effects of epithelial to mesenchymal transition on T cell targeting of melanoma cells. Frontiers in oncology, 4, p.367.
- Nusse, R. and Clevers, H., 2017. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell, 169(6), pp.985-999.
© 2023 - The Mathematical Oncology Blog