Discovering mechanisms of response via CSI (Cancer Systems Immunology)
Behind the paper
Cancer systems immunology reveals myeloid—T cell interactions and B cell activation mediate response to checkpoint inhibition in metastatic breast cancer
Edgar Gonzalez, Jesse Kreger, Yingtong Liu, Xiaojun Wu, Arianna Barbetta, Aaron G. Baugh, Batul Al-Zubeidy, Julie Jang, Sarah M. Shin, Matthew Jacobo, Vered Stearns, Roisin M. Connolly, Won Ho, Juliet Emamaullee, Adam L. MacLean, Evanthia T. Roussos Torres
Read the paperA major cause of death in patients with breast cancer is complications related to metastasis to distant organs. The tumor microenvironment (TME) in metastatic tumors is complex and distinct from primary tumors, comprising various cell types, including tumor cells, stromal cells, and immune cells that influence tumor progression and response to treatments. Immune checkpoint inhibitors (ICIs) target many of these cell types to reduce inhibitory signaling to T cells, which have proved effective against some solid tumor types, but are ineffective against breast cancer on their own [1]. Sensitization of the immune-suppressed TME to ICIs by drugs such as epigenetic modulators shows promise, but the mechanisms of sensitization are unknown. To investigate changes in the TME in breast cancer metastases induced by treatment, we believe it is necessary to combine experimental and clinical data with theory, using an interdisciplinary, computational cancer systems immunology approach [2, 3]. This methodology is vital to learn mechanisms driving successful immunotherapeutic responses in breast cancer and other previously unresponsive solid tumor types.
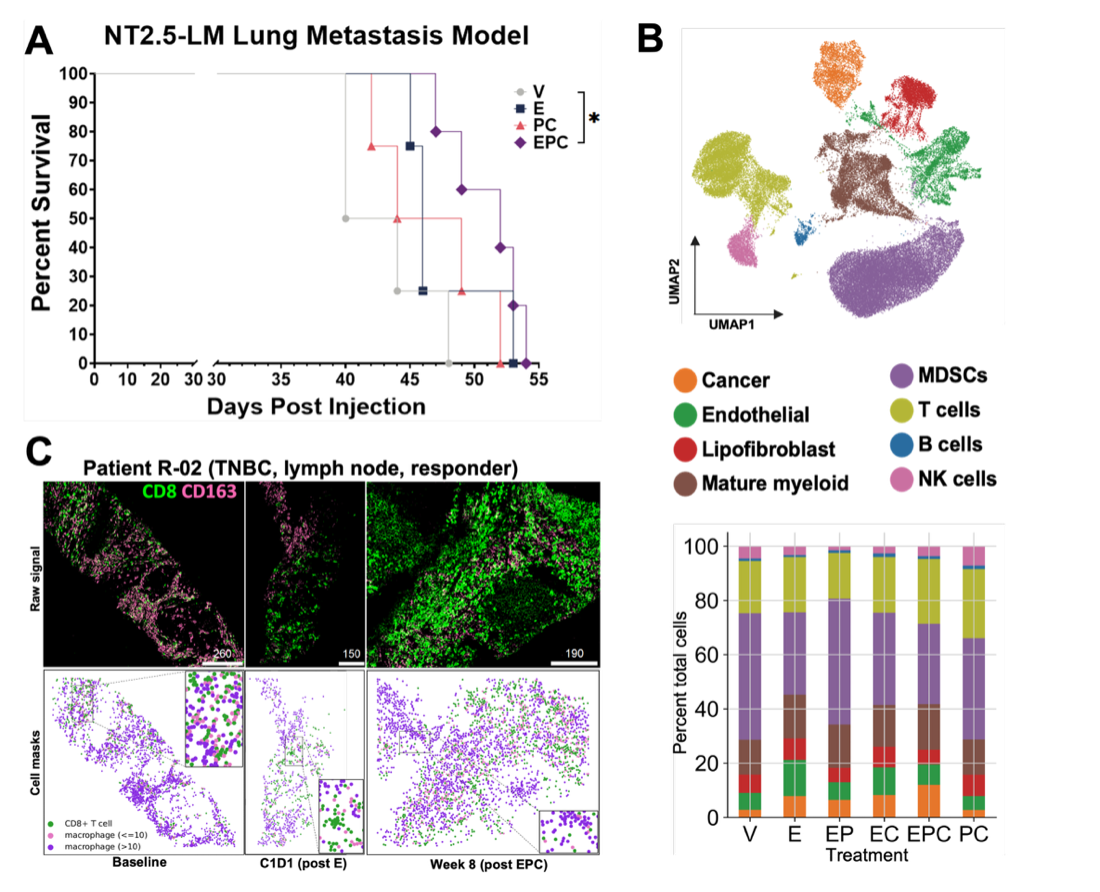
In our new preprint “Cancer systems immunology reveals myeloid---T cell interactions and B cell activation mediate response to checkpoint inhibition in metastatic breast cancer” [4] we integrated preclinical data, clinical data, and mathematical modeling to uncover the complex changes in immune cells and their interactions with other cells that are associated with an anti-tumor immune response in metastatic breast cancer (Fig. 1A). We present a comprehensive analysis of single-cell RNA-sequencing data (with 39 cell states!) from treated murine breast-to-lung metastases (Fig. 1B) as well as spatial proteomics from patient specimens from our clinical trial NCI-9844 (nivolumab + ipilimumab + entinostat) [5] (Fig. 1C). We identified interactions between myeloid-derived suppressor cells, tumor-associated macrophages, and T cells, in addition to changes in B cell activation, that are most influential in dictating responses to treatment with a histone deacetylase inhibitor (entinostat) in combination with dual checkpoint inhibition. We report significant changes in immune cell infiltration, signaling via ICAM-1 and interferon gamma, and an immunoglobulin class switch from IgA to IgG in murine and/or patient samples with combination treatment. Further validation of preclinical findings is highlighted by the presence of mature tertiary lymphoid structures and a decrease in macrophage–CD8+ T cell interactions in tumors from patients who responded to treatment.
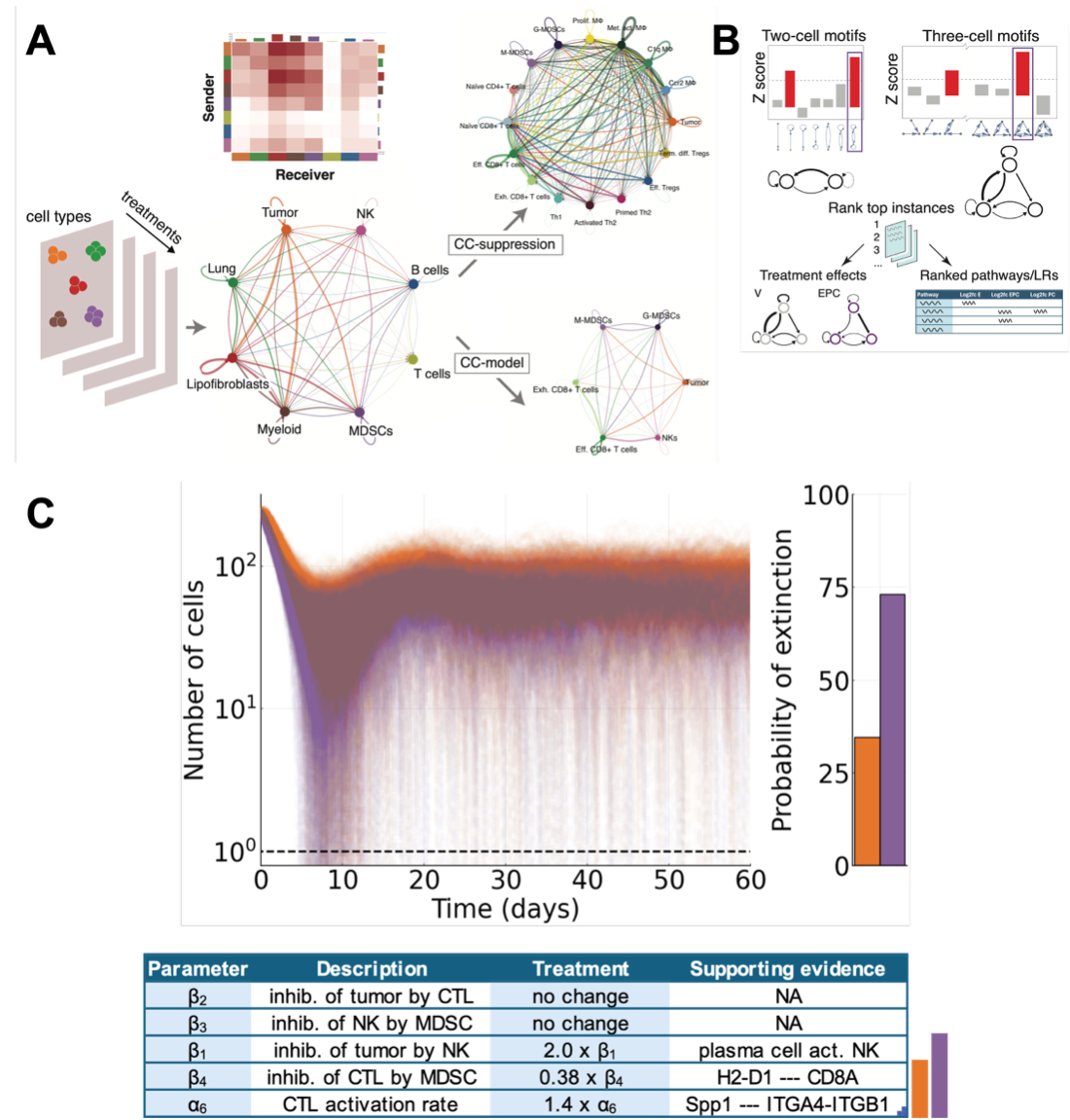
Through the combination of computational tools [6, 7] that infer cell-cell communication networks (Fig. 2A-B), and innovative mathematical modeling of anti-tumor immune responses informed by both preclinical and clinical data (Fig. 2C), we found the most important interactions in the TME that are required to reduce or eliminate metastatic tumors. We conclude that it is the carefully orchestrated set of changes in cells of lymphoid and myeloid origin, induced by epigenetic modulation, that sensitize the TME to checkpoint inhibition. These findings not only identify which cell types should be targeted in metastatic breast cancer but also underscore the immense potential for a cancer systems immunology approach to accelerate the discovery of mechanisms of response to novel therapeutics. Beyond the impact of the current mechanisms elucidated, this study serves as a framework for other investigations into mechanisms of response to new therapeutics in complex TMEs.
References
- Dvir, K., Giordano, S. & Leone, J. P. Immunotherapy in Breast Cancer. Int. J. Mol. Sci. 25, 7517 (2024). doi: 10.3390/ijms25147517
- Reticker-Flynn, N. E. & Engleman, E. G. Cancer systems immunology. eLife 9, e53839 (2020). doi: 10.7554/eLife.53839
- Way, G. P. et al. A field guide to cultivating computational biology. PLOS Biol. 19, e3001419 (2021). doi: 10.1371/journal.pbio.3001419
- Gonzalez, E. et al. Cancer systems immunology reveals myeloid - T cell interactions and B cell activation mediate response to checkpoint inhibition in metastatic breast cancer. (2025). doi: 10.1101/2025.06.09.658361
- Roussos Torres, E. T. et al. Entinostat, nivolumab and ipilimumab for women with advanced HER2-negative breast cancer: a phase Ib trial. Nat. Cancer 5, 866–879 (2024). doi: 10.1038/s43018-024-00729-w
- Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021). doi: 10.1038/s41467-021-21246-9
- Mayer, S. et al. The tumor microenvironment shows a hierarchy of cell-cell interactions dominated by fibroblasts. Nat. Commun. 14, 5810 (2023). doi: 10.1038/s41467-023-41518-w
© 2026 - The Mathematical Oncology Blog