A general deterministic framework for modeling chromosome missegregations
Behind the Paper
Intra-tumor heterogeneity, turnover rate and karyotype space shape susceptibility to missegregation-induced extinction
Gregory J. Kimmel, Richard J. Beck, Xiaoqing Yu, Thomas Veith, Samuel Bakhoum, Philipp M. Altrock, Noemi Andor
Read the paperModeling mis-segregations is hard because of the sheer number of possible karyotypes. More than 70 quintillions (considering 8 states/chr)! We derived a deterministic framework for modeling mis-segregations (MS) of multiple chromosome types accounting for viable karyotype intervals of all 22 autosomes simultaneously and for the turnover rate of the population [1]. It offers the flexibility to identify potential synergies between copy number changes of multiple chromosomes and to model intra-tumor heterogeneity in the copy number of all chromosomes, as well as karyotype specific MS- and turnover rates.
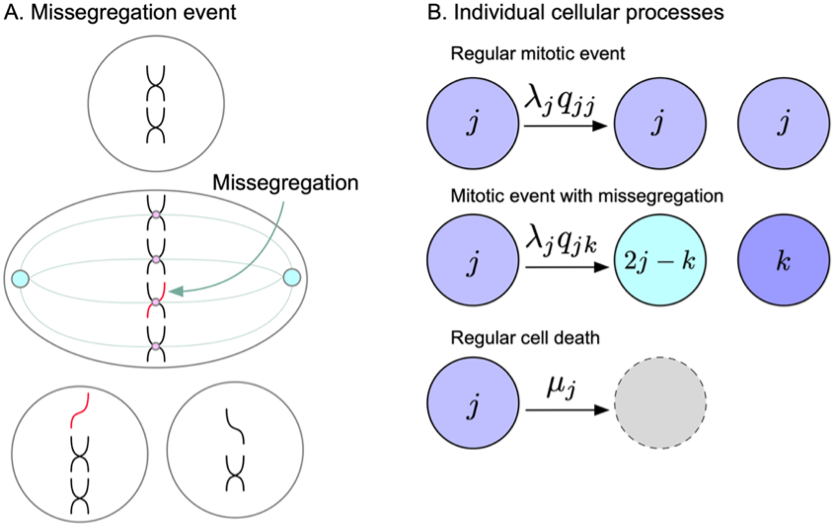
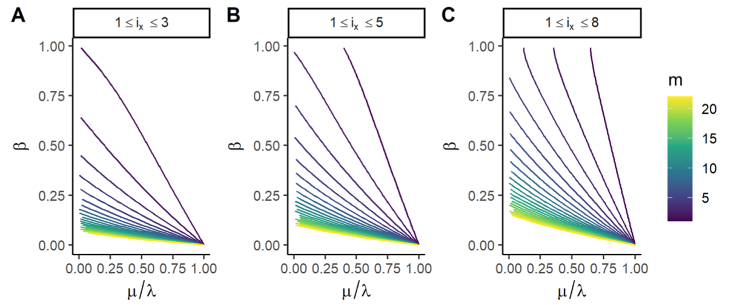
As first application of the model, we predict MIE – i.e. the maximum MS rate above which a population would go extinct due to frequent MS into non-viable copy number states. We found that heterogeneous populations evolved toward the karyotype with the lowest MS rate. Theoretical and numerical solutions converge on a necessary condition for MIE: that karyotypes with low MS rates must also have high death rates. This prediction supports empirical evidence of a convergence to genomic stability at advanced stages of tumor progression. Reduction in overall fitness for aneuploidy in many normal cells has been well described [2]. The mechanism for fitness reduction is driven largely through changes in gene expression. Aneuploidy causes gene-dosage imbalances resulting in metabolic changes and is often accompanied by p53-mediated cellular senescence [3, 4, 5].
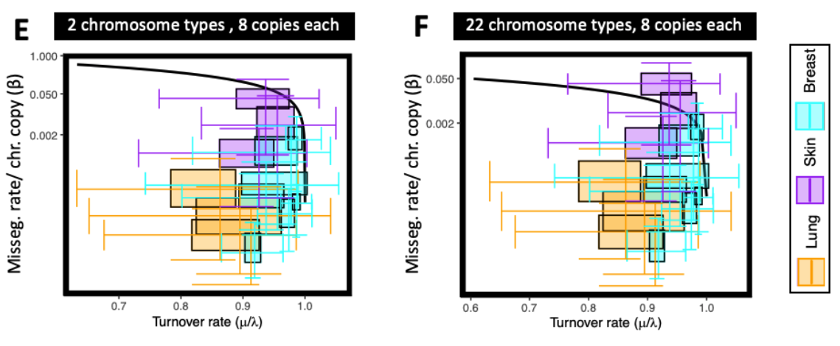
We leverage a finding reported in Bakhoum et al. [6], which identified IFG as the top pathway that can approximate chromosome MS. Using IFG as a proxy of MS, the majority of tumors had turnover- and MS rates that were within regions of parameter space predicted as viable.
Future applications of the model will take advantage of a key feature of our framework – its ability to model MS of multiple chromosome types. The phenotypic consequence of gaining or losing a copy of a given chromosome is a function of what other chromosomes are in that nucleus, leading to a combinatorial explosion of phenotypic possibilities. E.g. considering up to five copies per chromosome yields 522 possible copy number states for autosomes alone. Viability of specific trisomies likely depend on the genetic background or tissue of origin of a cell [7, 8]. For example, a study analyzing chromosome arm aneuploidies of 6,977 tumours identified 88 synergistically co-occurring aneuploidy pairs as multivariate prognostic biomarkers in 58% of patients [9]. Notably, 86% of these aneuploidies were individually no significant predictors, indicating two separate MS events must happen to facilitate this synergy [9]. Melanoma cells losing a copy of chromosome 8p showed reduced fitness, yet when the same cells also gained a copy of chromosome 6p they surpass fitness of cells with neither alteration. By accounting for viable karyotype intervals of all 22 autosomes simultaneously we expect our model to predict whether populations can reach global fitness peaks or whether extinction events at local fitness valleys will prevent them from doing so.
Another key feature of this framework is the option of modeling karyotype specific MS- and turnover rates. A daughter cell after a MS event will have a large fraction of its DNA in an altered copy number state. But for a MS event to change a cell's karyotype, it is not enough for the event to happen, the cell must also survive it. The standard fate of aneuploid cells generated from a MS event is to die during interphase. The sudden genome-dosage imbalance caused by a MS event [10] induces p53-mediated cellular senescence in the subsequent G1 phase [3, 4, 5]. While cancer cells evolve to avert MS-induced cell death more proficiently, a MS still activates p53 in the G1 phase, even among cancer cells5, albeit less reliably [11]. In particular, death is elevated in cells that missegregate higher numbers of chromosomes in a single division [12]. E.g. in a study lead by Santaguida et al. only 9% of cells that missegregated chromosomes were arrested in G1, while remaining 80 percent continued to divide [11]. What factors govern a cell's risk to die after an MS are still largely unknown. One contributing factor is what % of the genome has been altered in a missegregating cell. Single cell sequencing of cells arrested in the interphase immediately following the mitosis during which chromosome MS was induced, revealed genomic imbalances involving more than 20% of their genomes. In contrast, aneuploid cells that still divided harbored genomic imbalances, which involved less than 5% of their genomes [11].
Accounting for heterogeneity in MS and turnover rates can possible overcome shortcoming of prior models of chromosome MSs. One of these limitations is that given enough time, prior models will eventually converge onto the same near-triploid optimal karyotype [13], despite considerable variability among founder cells' karyotype state or WGD status. This does not align with experimental observations [14, [15]. Prior models do not explain the wide distributions of ploidies observed in-vitro and in-vivo [13, 16]. The placement of tumors within these ploidy distributions is of predictive and prognostic relevance [4, 17, 18, 19].
References
- Kimmel, G.J., Beck, R.J., Yu, X., Veith, T., Bakhoum, S., Altrock, P.M., and Andor, N. (2023). Intra-tumor heterogeneity, turnover rate and karyotype space shape susceptibility to missegregation-induced extinction. PLoS Comput. Biol. 19, e1010815. doi: 10.1371/journal.pcbi.1010815.
- Santaguida, S., and Amon, A. (2015). Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat. Rev. Mol. Cell Biol. 16(8), 473–485. doi: 10.1038/nrm4025.
- Vitale, M. (2013). Intratumor BRAFV600E heterogeneity and kinase inhibitors in the treatment of thyroid cancer: a call for participation. Thyroid Off. J. Am. Thyroid Assoc. 23, 517–519. doi: 10.1089/thy.2012.0614.
- Bakhoum, S.F., and Compton, D.A. (2012). Chromosomal instability and cancer: a complex relationship with therapeutic potential. J. Clin. Invest. 122, 1138–1143. doi: 10.1172/JCI59954.
- Thompson, S.L., and Compton, D.A. (2010). Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 188, 369–381. doi: 10.1083/jcb.200905057.
- Bakhoum, S.F., Ngo, B., Laughney, A.M., Cavallo, J.-A., Murphy, C.J., Ly, P., Shah, P., Sriram, R.K., Watkins, T.B.K., Taunk, N.K., et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. doi: 10.1038/nature25432.
- López, S., Lim, E.L., Horswell, S., Haase, K., Huebner, A., Dietzen, M., Mourikis, T.P., Watkins, T.B.K., Rowan, A., Dewhurst, S.M., et al. (2020). Interplay between whole-genome doubling and the accumulation of deleterious alterations in cancer evolution. Nat. Genet. 52, 283–293. doi: 10.1038/s41588-020-0584-7.
- Vasudevan, A., Baruah, P.S., Smith, J.C., Wang, Z., Sayles, N.M., Andrews, P., Kendall, J., Leu, J., Chunduri, N.K., Levy, D., et al. (2020). Single-Chromosomal Gains Can Function as Metastasis Suppressors and Promoters in Colon Cancer. Dev. Cell 52, 413-428.e6. doi: 10.1016/j.devcel.2020.01.034.
- Shukla, A., Nguyen, T.H.M., Moka, S.B., Ellis, J.J., Grady, J.P., Oey, H., Cristino, A.S., Khanna, K.K., Kroese, D.P., Krause, L., et al. (2020). Chromosome arm aneuploidies shape tumour evolution and drug response. Nat. Commun. 11, 449. doi: 10.1038/s41467-020-14286-0.
- Sheltzer, J.M., Ko, J.H., Replogle, J.M., Burgos, N.C.H., Chung, E.S., Meehl, C.M., Sayles, N.M., Passerini, V., Storchova, Z., and Amon, A. (2017). Single-chromosome Gains Commonly Function as Tumor Suppressors. Cancer Cell, 31(2), 240–255. doi: 10.1016/j.ccell.2016.12.004.
- Santaguida, S., Richardson, A., Iyer, D.R., M’Saad, O., Zasadil, L., Knouse, K.A., Wong, Y.L., Rhind, N., Desai, A., and Amon, A. (2017). Chromosome mis-segregation generates cell cycle-arrested cells with complex karyotypes that are eliminated by the immune system. Dev. Cell 41, 638-651.e5. doi: 10.1016/j.devcel.2017.05.022.
- Silk, A.D., Zasadil, L.M., Holland, A.J., Vitre, B., Cleveland, D.W., and Weaver, B.A. (2013). Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc. Natl. Acad. Sci. 110, E4134–E4141. doi: 10.1073/pnas.1317042110.
- Laughney, A.M., Elizalde, S., Genovese, G., and Bakhoum, S.F. (2015). Dynamics of Tumor Heterogeneity Derived from Clonal Karyotypic Evolution. Cell Rep. 12, 809–820. doi: 10.1016/j.celrep.2015.06.065.
- Ben-David, U., Siranosian, B., Ha, G., Tang, H., Oren, Y., Hinohara, K., Strathdee, C.A., Dempster, J., Lyons, N.J., Burns, R., et al. (2018). Genetic and transcriptional evolution alters cancer cell line drug response. Nature 560, 325–330. doi: 10.1038/s41586-018-0409-3.
- Ben-David, U., Ha, G., Tseng, Y.-Y., Greenwald, N.F., Oh, C., Shih, J., McFarland, J.M., Wong, B., Boehm, J.S., Beroukhim, R., et al. (2017). Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 49, 1567–1575. doi: 10.1038/ng.3967.
- Elizalde, S., Laughney, A.M., and Bakhoum, S.F. (2018). A Markov chain for numerical chromosomal instability in clonally expanding populations. PLoS Comput. Biol. 14, e1006447. doi: 10.1371/journal.pcbi.1006447.
- Brastianos, P.K., Carter, S.L., Santagata, S., Cahill, D.P., Taylor-Weiner, A., Jones, R.T., Van Allen, E.M., Lawrence, M.S., Horowitz, P.M., Cibulskis, K., et al. (2015). Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 5, 1164–1177. doi: 10.1158/2159-8290.CD-15-0369.
- Angelova, M., Mlecnik, B., Vasaturo, A., Bindea, G., Fredriksen, T., Lafontaine, L., Buttard, B., Morgand, E., Bruni, D., Jouret-Mourin, A., et al. (2018). Evolution of Metastases in Space and Time under Immune Selection. Cell 175, 751-765.e16. doi: 10.1016/j.cell.2018.09.018.
- Kimmel, G.J., Dane, M., Heiser, L.M., Altrock, P.M., and Andor, N. (2020). Integrating mathematical modeling with high throughput imaging explains how polyploid populations behave in nutrient-sparse environments. Cancer Res. doi: 10.1158/0008-5472.CAN-20-1231.
© 2026 - The Mathematical Oncology Blog