Use this cell grammar to simplify systems biology modeling
Behind the paper
Human interpretable grammar encodes multicellular systems biology models to democratize virtual cell laboratories
Jeanette Johnson et al.
Read the paperOne of the foundational goals of systems biology is to predict the impact of cellular ecosystems on disease progression and therapeutic outcomes. Data-driven techniques from single-cell and spatial multi-omics have unlocked our ability to characterize these systems at single snapshots in time. However, making predictions forward in time from these measurements is challenged by the complexity of multicellular interactions and their evolution. Mathematical modeling techniques, including agent-based models (ABMs), allow us to encode multicellular interactions and mathematically predict the future of cancer ecosystems. In principle, embedding multi-omics data into ABMs provides the opportunity to make predictions from individual multicellular systems. Despite being well suited to many systems biology questions, the coding and mathematical expertise required to create ABMs have thus far limited their broad application to represent diverse biological systems or seamlessly integrate experimental data. Moreover, because most ABMs tend to be hand-coded (“artisinal handcrafted C++”), it is difficult to maintain, share, and reuse them, substantially reducing their impact and reproducibility.
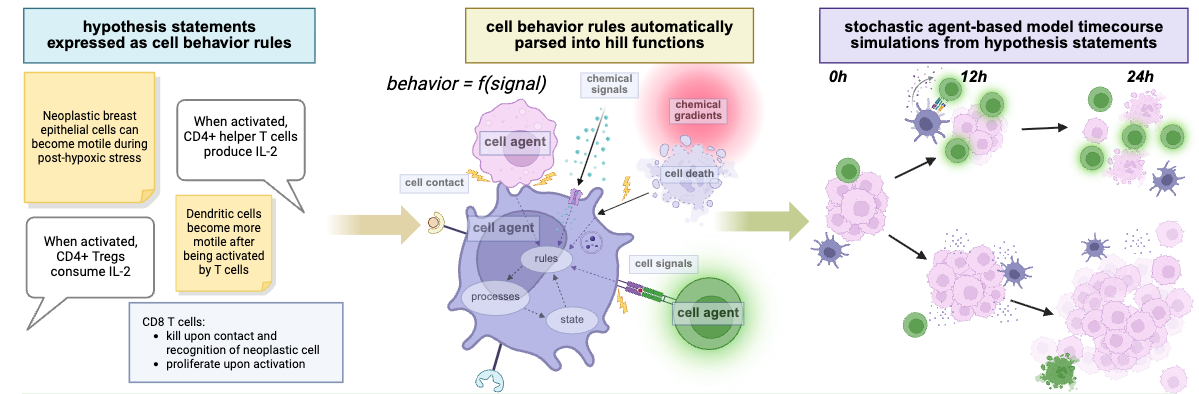
To address these challenges, we have been working to simplify the process of creating these models. One of the first steps is to identify and unify the commonalities in currently used mathematical models of cancer and tissue ecosystems. Our new paper (Johnson et al., Cell (2025))[1] is a critical step in that direction, introducing flexible cell behavior grammar capable of representing hypotheses of cell behavior in human-readable statements which can then be automatically applied to define agent behavior. Observational statements like "in macrophages, IL-6 increases migration speed" along with a “chemotactic sensitivity to IL-6” can be read by PhysiCell and parsed into mathematics and code at run time, yielding a model that causes macrophage agents to migrate toward their substrate of choice at a faster rate when the simulated IL-6 substrate concentration in the local environment increases. The signal thresholds and saturation points that modify behavior are expressed directly within the grammar, so each response curve is independently tuneable using only these single-line rule statements. This significantly cuts down on the startup time for developing any new PhysiCell model, and simplifies and speeds up the process of developing the model system, while also making it simpler and faster to interactively add and remove rules.
Model rules can be written in plain language, sourcing information by experts, literature mining, or direct data analysis. New rules can augment old rules without breaking them, allowing us to use these models to aggregate, refine and share knowledge over time. We hope this tool can help open up "virtual laboratories" within real laboratories, for use in virtual thought experiments.
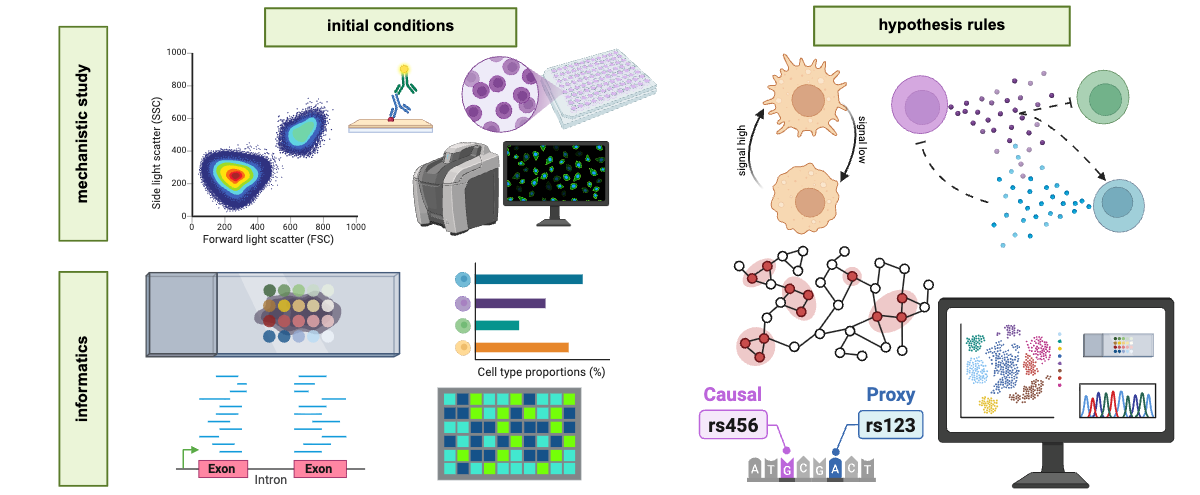
Our manuscript shows a series of examples using the new grammar, including re-writing prior modeling work in Rocha et al., iScience (2021) where breast cancer cells respond to low oxygen by increasing invasion, and phenotypic persistence allows cancer cells to form “plumes” that break through normoxic zones to escape the tumor. This model can be written in minutes using only a user-friendly GUI called PhysiCell Studio and rules written in our new grammar.
We worked closely with collaborators at OHSU to write a model of around 12 simple rules describing an immune response to a hypoxic tumor, including eventual immune exhaustion. This model can be built entirely with the new grammar, with zero extra C++. Moreover, even though the rules were written and developed in 2D, the same rules (with very minor change to a pressure-based rule) can be used in 3D after changing initial conditions. Here’s a sample video of the dynamics.
Once our system was benchmarked to simulate tumor growth through immune interactions, we were able to extend this model to simulate the seemingly counterintuitive role of T cells on promoting tumor invasion through signaling from macrophages observed previously in DeNardo et al., Cancer Cell (2009). The model enabled us to predict that the invasive role associated with EGFR signaling in that paper likely arose from enhancing tumor cell motility, which we validated experimentally. This example demonstrated how the modeling framework can motivate and focus new experiments to validate the mechanisms underlying predicted cell behavior.
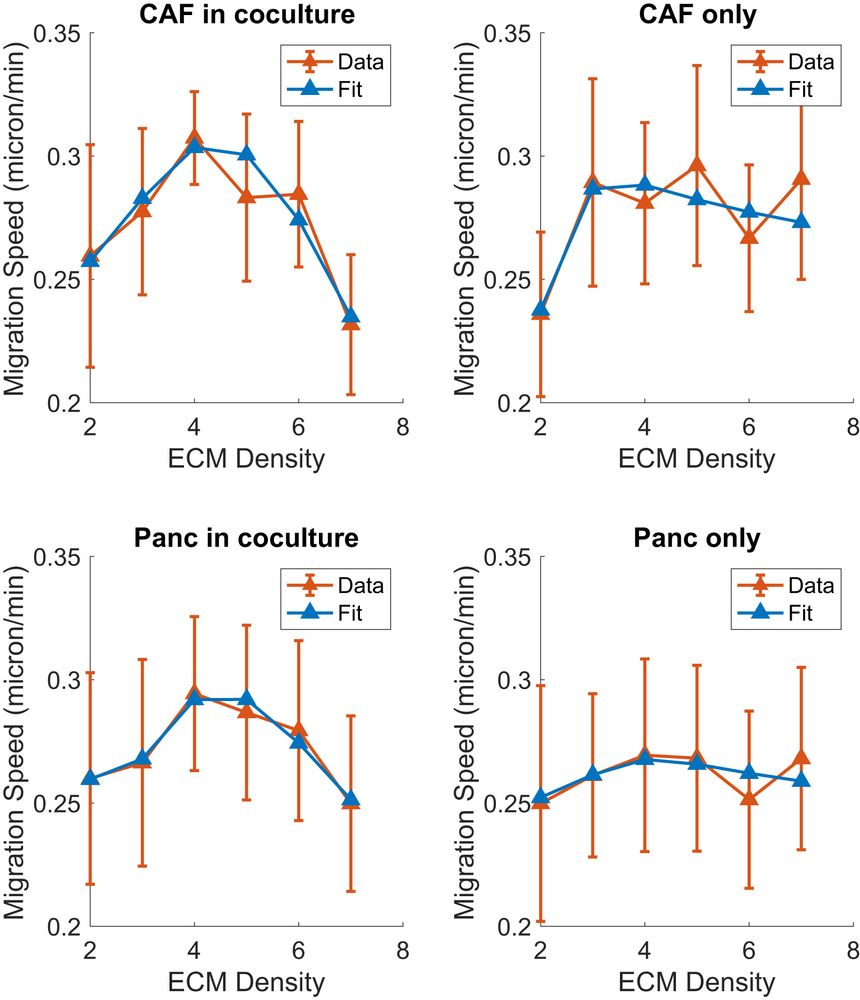
Digging deeper, we worked with a wide network of collaborators to apply the grammar to create a model of pancreatic cancer (PDAC). Previously, our high-throughput genomics studies showed that cancer-associated fibroblasts (CAFs) induce epithelial to mesenchymal transition (EMT) in tumor cells, a phenotype that we observed is mutually exclusive from cell proliferation. We sought to input this into the hypothesis grammar to see if our model could thereby explain the dual tumor-promoting and tumor-suppressing role of CAFs in PDAC. However, the precise cellular behavior from EMT and the impact of the extracellular matrix (ECM) remained unknown from genomics, requiring further parameterization for our models. The relationships between ECM density and cell migration were revealed by live cell imaging experiments at Johns Hopkins, and these data were integrated directly to inform the model using a set of rules describing how the ECM both inhibits and drives motility depending on the cell type and concentration.
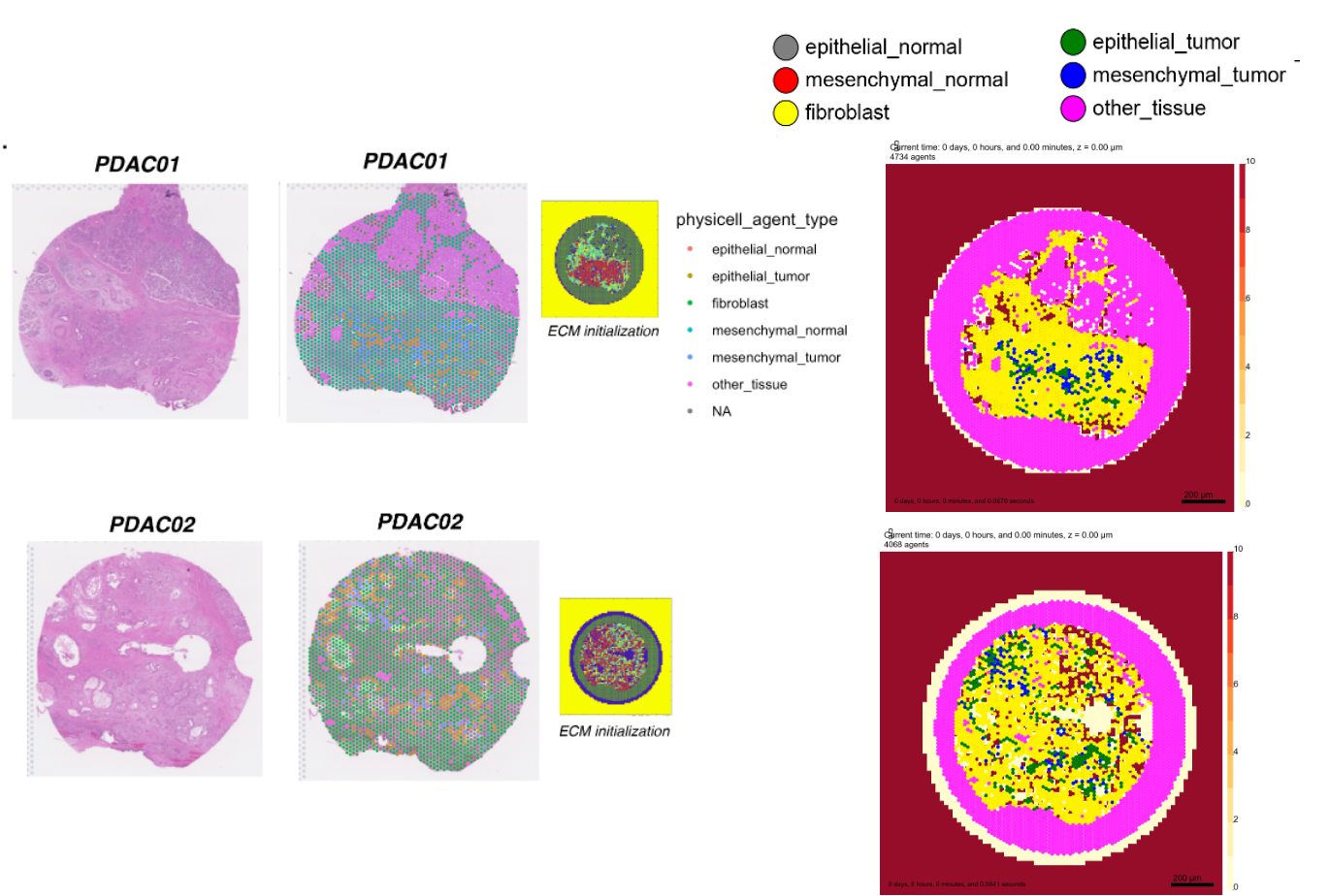
We were able to define ABM cell identities, positions, and even cell rules directly from our spatial transcriptomics data of PDAC (single-cell gene
sequencing that preserves cell position), and we directly initialized models from an individual tissue’s
cellular composition. This showed how cancer-fibroblast interactions can drive malignant transformation (EMT), but in
some cases, these interactions can also trap cancer cells to prevent spread. Model simulations suggest that the
cellular motility induced by EMT supports tumor cell spread to regions that are CAF poor, where they are free to
proliferate and invade. These observations are consistent with the counterintuitive limited clinical benefit of
CAF-inhibiting monotherapies and unanticipated subtype switching observed in PDAC metastasis in other experimental
and clinical studies.

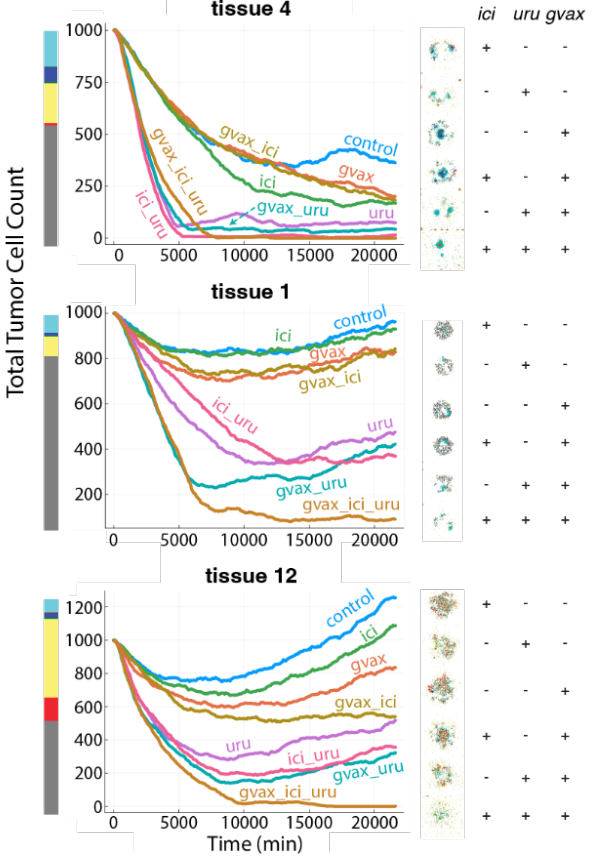
We also used the model to create a "virtual laboratory" to test combination immunotherapies based upon a
real world platform, combination immunotherapy clinical trial in pancreatic cancer (Li et al., Canc. Cell (2022), Heumann et al., Nat. Comm. (2023)). Matching initial
immune cell abundances from pre-treatment cellular compositions of individual patients in a reference single-cell dataset of untreated PDAC enabled us
to individualize microenvironment conditions and predict their impact on therapeutic response. Combining population
shifts that encode the effects of targeted immunotherapies allowed us to simulate and predict combination therapies,
with significant differences in ranked efficacy based on initial immune microenvironment profiles.

We note that the hypothesis grammar and agent-based models can apply to all multicellular biological systems that
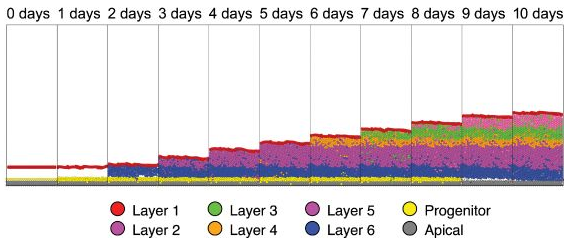
change in time. Here, we show that the grammar extends beyond representing cancer or cancer immunology systems. We
also built a model using the Allen Institute Brain Atlas data, and modeled neurocortical development, obtaining
rules that generate the different layer structures responsible for different types of cognition. In this example,
the spatial transcriptomics served as the goal for the model. The parameters driving the rules were then tuned to
show how the same rules operating with context specific modifications can generate distinct regions of the cortex,
in this case the motor and somatosensory cortexes. This reflects how the body uses the same rules in different ways
to organize cells into tissue structures specialized for
specific functions.[a]
We hope this framework and cell behavior grammar will make ABMs a relevant and feasible way to represent important cancer systems and many other multicellular systems, and will facilitate their adoption by a wider range of mathematicians and biologists at a range of career stages. In the near future, we envision that model rules using this grammar will be readily shareable in “public libraries” of cell definitions, and we are already building cloud-based versions of PhysiCell on Galaxy and NanoHUB that can be run without any software installations. We encourage you to explore tutorial materials, and maybe even attend a future virtual hackathon!
It was a great honor to work with this team, and we're beyond grateful for funding support from the National Institutes of Health (NIH), including notably the National Cancer Institute’s Informatics Technology for Cancer Research (ITCR) Program and Human Tumor Atlas Network (HTAN), the NIH BRAIN Initiative, the Jayne Koskinas Ted Giovanis Foundation for Health and Policy, the National Foundation for Cancer Research, Break Through Cancer, and the Lustgarten Foundation. Their generous support and grants for basic science research, team science, software infrastructure, and risky ideas positioned us to create this work today.
References
- J.A.I. Johnson, D.R. Bergman, H.L. Rocha, D.L. Zhou, E. Cramer, I.C. Mclean, Y.W. Dance, M. Booth, Z. Nicholas, T. Lopez-Vidal, A. Deshpande, R. Heiland, E. Bucher, F. Shojaeian, M. Dunworth, A. Forjaz, M. Getz, I. Godet, F. Kurtoglu, M. Lyman, J. Metzcar, J.T. Mitchell, A. Raddatz, J. Solorzano, A. Sundus, Y. Wang, D.G. DeNardo, A.J. Ewald, D.M. Gilkes, L.T. Kagohara, A.L. Kiemen, E.D. Thompson, D. Wirtz, L.D. Wood, P.-H. Wu, N. Zaidi, L. Zheng, J.W. Zimmerman, J.M. Phillip, E.M. Jaffee, J.W. Gray, L.M. Coussens, Y.H. Chang, L.M. Heiser, G.L. Stein-O’Brien, E.J. Fertig, and P. Macklin, Human interpretable grammar encodes multicellular systems biology models to democratize virtual cell laboratories, Cell 188(17): 4711-4733.e37 (2025). doi: 10.1016/j.cell.2025.06.048
© 2026 - The Mathematical Oncology Blog