Mathematical model of colorectal cancer initiation
Behind the paper
Mathematical model of colorectal cancer initiation
Chay Paterson, Hans Clevers, Ivana Bozic
Read the paper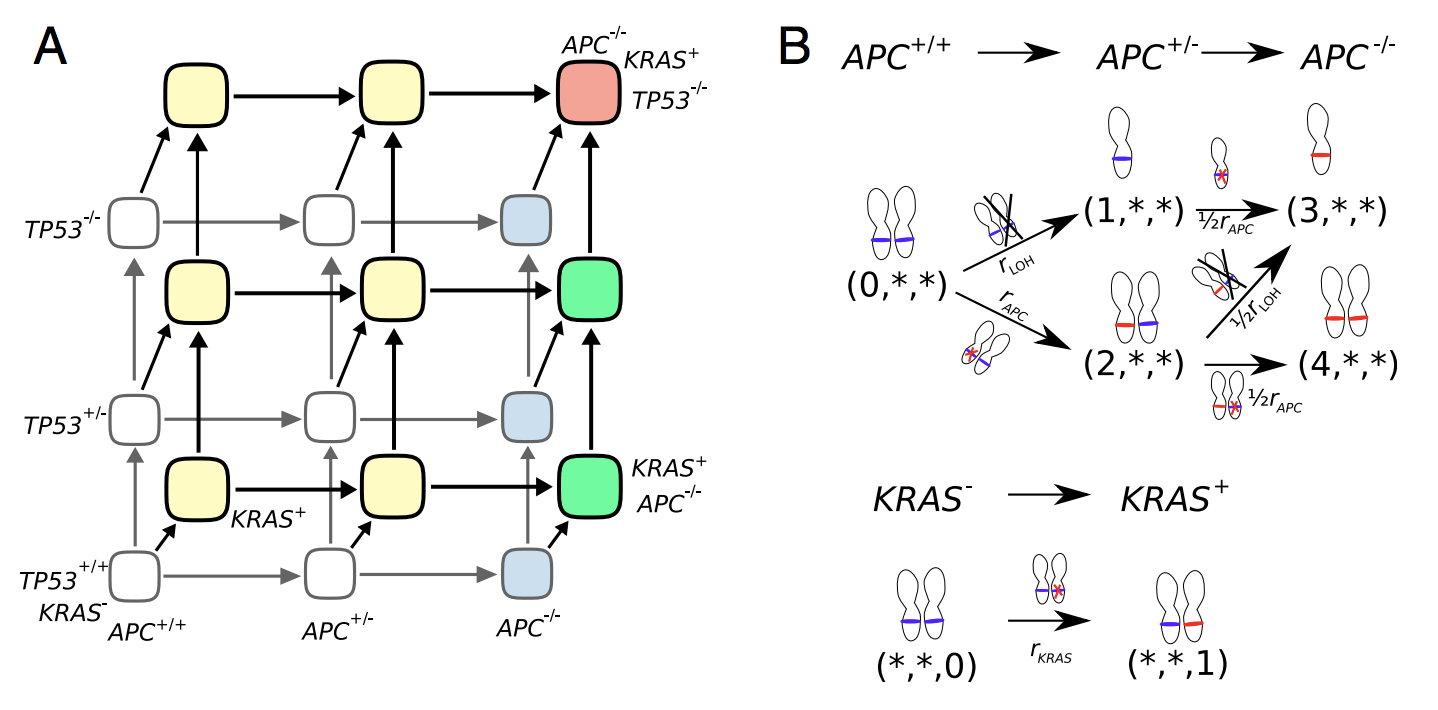
Figure 1. (A) Schematic of colorectal cancer initiation. Healthy (wild type) crypt is in the lower left corner, and a fully malignant crypt in the top right. (B) Top, transition rates from APC-wild type genotype (0,*,*) to fully inactivated APC through LOH and mutation (or vice versa) (3,*,*) or double mutation (4,*,*). Bottom, transition rate from wild-type KRAS (*,*,0) to activated KRAS (*,*,1).
We started with a simpler model in which the three driver events accumulate neutrally and showed that the neutral assumption leads to colorectal cancer (CRC) incidence that is many orders of magnitude lower than reported. Then we tackled the full model, in which we allowed colorectal crypts with driver mutations to divide according to a stochastic birth process. We were lucky that two groups measured the rates of expansion of colorectal crypts containing APC and KRAS mutations in human subjects in vivo3,4, and that another group showed that TP53 may not be providing growth advantage on its own5, so we were able to parametrize the model fully. Strikingly, we found that the reported lifetime risk of colorectal cancer can be recovered using our mathematical model of CRC initiation together with experimentally measured mutation rates in colorectal tissues and proliferation rates of premalignant lesions. One of the most interesting things we discovered is that the most likely order in which driver mutations are accumulated on the way to CRC in our mathematical model is APC-KRAS-TP53, exactly the same ordering that has been reported experimentally2. Not only that, we found that the most likely order of driver mutations is one that maximizes fitness along the carcinogenic path, so the driver mutation that provides maximum fitness advantage over healthy tissue is collected first and so on. We also realized that the question of mutation order is a bit subtle, and that one should distinguish between the order in which driver mutations are accumulated in typical crypts (large majority of which will not end up cancerous) and the most likely order of driver mutations that will result in cancer within a patient’s lifetime. For example, we find that healthy crypts will typically first get an activating mutation in KRAS, but crypts that will become cancerous will typically first inactivate APC. In sum, our detailed genetic model contributes to the quantitative understanding of the process of colorectal carcinogenesis in patients; in the future it could be extended to include other significant driver genes, and to allow the mutation rate and fitness advantage provided by drivers to vary between patients and throughout the patients’ lifetimes, reflecting differences in genetic background and the tumor microenvironment6.References
- F. Blokzijl et al., Tissue- specific mutation accumulation in human adult stem cells during life. Nature 538, 260-264 (2016).
- B. Vogelstein et al., Genetic alterations during colorectal-tumor development. N Engl J Med 319, 525-532 (1988).
- A. M. Baker et al., Quantification of crypt and stem cell evolution in the normal and neoplastic human colon. Cell Rep. 8, 940-947 (2014).
- A. M. Nicholson et al., Fixation and spread of somatic mutations in adult human colonic epithelium. Cell Stem Cell 22, 909-918 (2018).
- L. Vermeulen et al., Defining stem cell dynamics in models of intestinal tumor initiation. Science 342, 995-998 (2013).
- Fane, M. & Weeraratna, A. T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 20, 89–106 (2020).
© 2026 - The Mathematical Oncology Blog