Optimal dosing of anti-cancer drug treatment under drug-induced plasticity
Behind the paper
Optimal dosing of anti-cancer treatment under drug-induced plasticity
Einar Bjarki Gunnarsson, Benedikt Vilji Magnússon, Jasmine Foo
Read the paperBackground
While cancer has traditionally been considered a genetic disease, mounting evidence indicates an important role for non-genetic (epigenetic) mechanisms like DNA methylation and histone acetylation, which influence gene expression without changing the genetic code (Figure 1) [1, 2, 3, 4]. Several experimental works have shown that these reversible mechanisms can enable tumor cells to temporarily persist drug treatment and set the stage for the evolution of more permanent resistance mechanisms [5, 6, 7, 8]. In a previous work, we studied a mathematical model of drug resistance involving both genetic and non-genetic mechanisms [9]. We showed that the ability to adopt transient non-genetic drug tolerance can be sufficient to drive tumor recurrence, even in the absence of any genetic events. This may help explain why many tumors seem to develop drug resistance without acquiring mutations in drug targets [10].
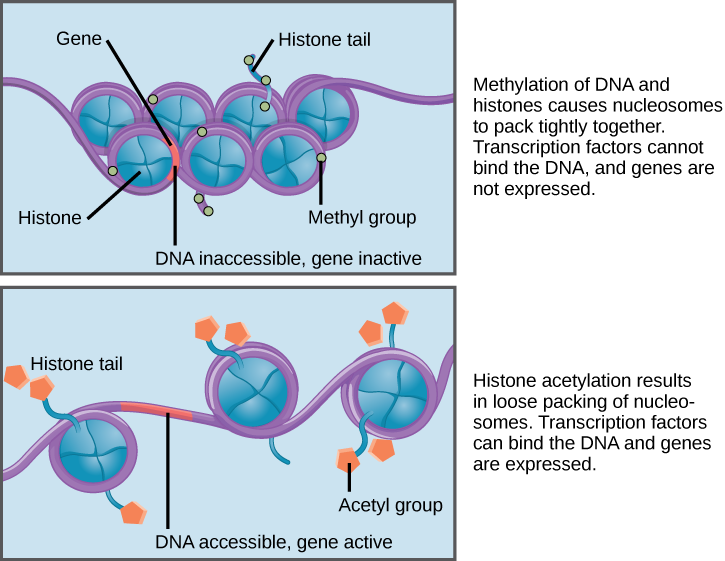
More recent experimental evidence indicates that the adoption of non-genetic drug tolerance can be directly induced by the anti-cancer drug [8, 11, 12, 13]. This confounds conventional maximum tolerated dose (MTD) strategies aimed at eradicating the tumor bulk, since it creates a trade-off between killing the tumor bulk and accelerating the evolution of resistance. It is therefore necessary to develop dosing protocols that appropriately balance this trade-off. We are excited to introduce our latest preprint which tackles this important problem [14].
Mathematical model
We consider a mathematical model of reversible drug tolerance where tumor cells are able to transition between two cell states, a drug-sensitive (type-0) and a drug-tolerant (type-1) state (Figure 2(a)). For simplicity, the anti-cancer drug is assumed to increase the death rate of sensitive cells (cytotoxic drug), but our results generalize to cytostatic drugs which lower the division rate of sensitive cells (Figure 2(b)). Furthermore, to model drug-induced tolerance, the transition rate from sensitivity to tolerance is assumed to either increase linearly as a function of the drug dose (linear induction of tolerance) or to increase uniformly whenever the drug is present (uniform induction of tolerance) (Figure 2(c)). These two forms of drug-induced tolerance were observed in a recent experimental investigation of colorectal cancer [8].
From a biological perspective, it is equally reasonable that the drug facilitates drug tolerance by inhibiting transitions from tolerance to sensitivity, or that the drug affects both transitions between sensitivity and tolerance simultaneously [12, 13, 15]. Therefore, we also allow the transition rate from tolerance to sensitivity to be linearly decreasing in the drug dose or uniformly reduced whenever the drug is present (Figure 2(d)). With these assumptions, we incorporate several different potential forms of drug-induced tolerance, and one of our primary goals is to understand how these different potential forms influence the optimal dosing strategy.

Main results
We begin by considering linear induction of tolerance. We study three cases: In Case I, the drug only elevates transitions from sensitivity to tolerance, in Case II, the drug only inhibits transitions back, and in Case III, it has both effects simultaneously (Figure 3).
In all cases, the optimal dosing strategy involves both a transient phase and an equilibrium phase (Figure 3, top row). During the equilibrium phase, a constant dose $c^\ast$ is applied, which maintains the tumor at a fixed proportion between sensitive and tolerant cells (Figure 3, middle row). Therefore, we can say that the goal of the optimal dosing strategy is to steer the tumor to a desired intratumor composition and maintain it there. We can prove mathematically that the optimal equilibrium dose $c^\ast$ minimizes the long-run tumor growth rate $\sigma(c)$ under a constant dose $c$, given by the explicit expression $$ \sigma(c) = \frac{1}{2} \Big(\lambda_0(c)-\mu(c) + \lambda_1-\nu(c) + \sqrt{(\lambda_0(c)-\mu(c)-\lambda_1+\nu(c))^2+4\mu(c)\nu(c)}\Big). \qquad (\text{1}) $$
This implies that expression (1) has clinical value on its own, since it can be used to address questions concerning ultimate treatment success. For example, if we are interested in knowing whether there exists any dosing schedule (however complicated) capable of driving long-run tumor reduction, we simply need to check whether there exists a constant dose $c$ such that $\sigma(c) \lt 0$.
Under Case I, it is optimal to apply the constant dose $c^\ast$ from the beginning of treatment, while under Case II, it is optimal to start with larger doses during the transient phase (Figure 3, top row). This is because the tumor starts out as primarily sensitive, and under Case I, a high initial dose will induce a large number of sensitive cells to adopt tolerance. In the bottom row of Figure 3, we compare tumor size evolution under the optimal strategy vs. the constant-dose strategy applying $c^\ast$ throughout, which confirms the usefulness of applying a different dosing strategy during the transient phase than the equilibrium phase (for Cases II and III).

We then consider uniform induction of tolerance. Just like for the linear case, the optimal dosing strategy involves both a transient and an equilibrium phase, but a low constant dose is no longer optimal in the long run. Under Case I, it is optimal to apply the MTD continuously throughout treatment. Under Cases II and III, it is optimal to apply the MTD initially and to then alternate arbitrarily quickly between the MTD and no dose. Examples of optimal strategies are shown in the upper row of Figure 4, where the proportion of drug exposure over time is shown, assuming arbitrarily fast alternation between the MTD and no dose. The lower row of Figure 4 shows that just like for the linear case, the optimal dosing strategy maintains the tumor at a fixed proportion between sensitive and tolerant cells, implying that it remains true that the goal of the optimal treatment is to steer the tumor to a desired (optimal) long-run composition.

Conclusion and future directions: Personalizing treatment
Our work shows that the optimal dosing strategy depends heavily on the dynamics of drug-induced tolerance. Depending on the dynamics, it can be optimal to apply a low dose continuously, to apply the MTD continuously (implying that drug-induced tolerance does not in and of itself mean that MTD treatment is suboptimal), start with the MTD and then apply a low dose continuously, or start with the MTD and then alternate between the MTD and no dose.
Given the widespread genetic and epigenetic heterogeneity between patients, even for a given cancer type, our results indicate the need to better understand the dynamics of drug-induced tolerance on a patient-by-patient basis. A future ambition of ours is to help develop a pipeline integrating mathematical modeling with experiments on patient-derived tumor samples, where the goal is to infer a mathematical model of drug-induced tolerance and derive optimal model-informed dosing strategies on an individual patient basis.
We invite everyone to join a conversation on this topic in the mini-symposium “Decoding drug-induced persistence: Experiments, Models, and Optimal Drug Scheduling” (co-organized with Maximilian Strobl) at the upcoming 2025 SMB annual conference in Edmonton, Canada.
References
- R Brown and G Strathdee. Epigenomics and epigenetic therapy of cancer. Trends Mol Med, 8(4):S43–S48, 2002. DOI: 10.1016/S1471-4914(02)02314-6.
- PA Jones and SB Baylin. The epigenomics of cancer. Cell, 128(4):683–692, 2007. DOI: 10.1016/j.cell.2007.01.029.
- A Brock, H Chang, and S Huang. Non-genetic heterogeneity - a mutation-independent driving force for the somatic evolution of tumours. Nat Rev Genet, 10(5):336, 2009. DOI: 10.1038/nrg2556.
- WA Flavahan, E Gaskell, and BE Bernstein. Epigenetic plasticity and the hallmarks of cancer. Science, 357(6348):eaal2380, 2017. DOI: 10.1126/science.aal2380.
- SV Sharma, DY Lee, B Li, MP Quinlan, F Takahashi, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell, 141(1):69–80, 2010. DOI: 10.1016/j.cell.2010.02.027.
- AN Hata, MJ Niederst, HL Archibald, M Gomez-Caraballo, FM Siddiqui, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med, 22(3):262–269, 2016. DOI: 10.1038/nm.4040.
- SM Shaffer, MC Dunagin, SR Torborg, EA Torre, B Emert, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature, 546(7658):431, 2017. DOI: 10.1038/nature22794.
- M Russo, S Pompei, A Sogari, M Corigliano, G Crisafulli, et al. A modified fluctuation-test framework characterizes the population dynamics and mutation rate of colorectal cancer persister cells. Nat Genet, 54(7):976–984, 2022. DOI: 10.1038/s41588-022-01105-z.
- EB Gunnarsson, S De, K Leder, and J Foo. Understanding the role of phenotypic switching in cancer drug resistance. J Theor Biol, 490:110162, 2020. DOI: 10.1016/j.jtbi.2020.110162.
- RH Wilting and JH Dannenberg. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist Updat, 15(1-2):21–38, 2012. DOI: 10.1016/j.drup.2012.01.008.
- AO Pisco, A Brock, J Zhou, A Moor, M Mojtahedi, et al.. Non-darwinian dynamics in therapy-induced cancer drug resistance. Nat Commun, 4:2467, 2013. DOI: 10.1038/ncomms3467.
- A Goldman, B Majumder, A Dhawan, S Ravi, D Goldman, et al.. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat Commun, 6:6139, 2015. DOI: 10.1038/ncomms7139.
- Y Su, W Wei, L Robert, M Xue, J Tsoi, et al. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc Natl Acad Sci USA, 114(52):13679–13684, 2017. DOI: 10.1073/pnas.1712064115.
- EB Gunnarsson, BV Magnússon, and J Foo. Optimal dosing of anti-cancer treatment under drug-induced plasticity. arXiv preprint arXiv:2412.16391, 2024. DOI: 10.48550/arXiv.2412.16391.
- K Vipparthi, K Hari, P Chakraborty, S Ghosh, A Kumar, et al. Emergence of hybrid states of stem-like cancer cells correlates with poor prognosis in oral cancer. Iscience, 25(5), 2022. DOI: 10.1016/j.isci.2022.104317.
© 2026 - The Mathematical Oncology Blog