Exploiting evolutionary steering to induce collateral drug sensitivity in cancer
Behind the Paper
Exploiting evolutionary steering to induce collateral drug sensitivity in cancer
Ahmet Acar, Daniel Nichol, Javier Fernandez-Mateos, George D. Cresswell, Iros Barozzi, Sung Pil Hong, Nicholas Trahearn, Inmaculada Spiteri, Mark Stubbs, Rosemary Burke, Adam Stewart, Giulio Caravagna, Benjamin Werner, Georgios Vlachogiannis, Carlo C. Maley, Luca Magnani, Nicola Valeri, Udai Banerji, Andrea Sottoriva
Read the manuscript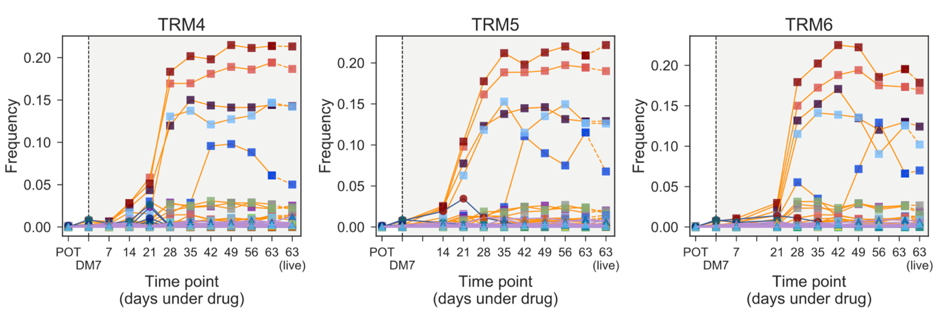
Figure 1: Temporal frequencies of the floating barcodes in three independent evolutionary replicates under exposure to the MEK inhibitor trametinib. Coloured squares indicate the frequency of unique barcodes. Barcode frequencies showed clear temporal dynamics, conserved evolutionary dynamics between replicates, and a close correspondence between the final floating barcode frequencies and associated frequencies of barcodes deriving from harvested live cells at the final time point.
To track this evolution at weekly time points we relied on floating DNA present in the cell culture media. We found that barcodes could be extracted from the milieu of cell-free DNA comprising DNA released by dying cells. These barcodes where then sequenced to provide weekly measurements of clonal composition. This seems paradoxical at first, why should the dead cells give information about resistant subclones? The answer lies in the population dynamics. As resistant subclones expand in frequency, cell-free DNA arising from the natural turnover of resistant cells outnumbers that from dying drug-sensitive cells that are diminishing in frequency. The results are striking (Figure 1). We see the same barcode-tagged resistant subclones arising between replicates, with very similar temporal dynamics. Furthermore, the final time point of media-derived barcode frequencies closely matches that sequenced from the live cells, confirming that the media-derived barcode frequencies serve as an accurate proxy of the live population composition. These results demonstrate repeatable evolutionary steering of a genetically heterogeneous cancer cell-line. The capacity to track clonal frequencies through time also suggests that this experimental system may prove well suited to the in vitro study of adaptive therapy10, or cancer evolutionary game theory11, where the dynamics of relative clonal frequencies are critical. Having demonstrated evolutionary steering, we sought to identify second-line drug collateral sensitivities through two contrasting approaches. First, we employed single-cell molecular profiling to help identify emergent molecular characteristics that might be exploited to yield collateral sensitivity. Second, we collaborated with colleagues in the CRUK Cancer Therapeutics Unit at the ICR to perform a high-throughput screen of 485 second-line compounds. Both approaches identified potential collaterally sensitive follow-ups for gefitinib or trametinib. Taken together our results indicate the potential for repeatable evolutionary steering towards collateral sensitivity in highly heterogeneous cancer populations. The robustness of these collaterally sensitive drug combinations will need be validated through further experiments. This work relied on an untested combination of large-population cell culture without serial passaging, high concentration drug exposures, lentiviral barcoding and sequencing, and DNA extraction from cell culture media. As such, we faced uncertainty about the validity of a potentially long and expensive experimental design and needed confidence in our approach from the outset. This confidence was found through a closely integrated mathematical modelling and wet lab biology approach. Each step of the experiment design, including the lentiviral barcoding process, population expansions, splitting into evolutionary replicates, and drug concentration calculations, was first validated in silico by mirroring the experimental through simulation with mathematical and computational models. This process gave us confidence that the clonal composition could be accurately identified from the DNA sequencing of barcodes if our hypotheses were indeed correct. Ultimately, this study demonstrates that close integration of mathematical modelling and experimental biology can greatly improve experimental design and yield results that either field alone would struggle to generate.References
- Greaves, Mel, and Carlo C. Maley. "Clonal evolution in cancer." Nature 481.7381 (2012): 306.
- Gillies, Robert J., Daniel Verduzco, and Robert A. Gatenby. "Evolutionary dynamics of carcinogenesis and why targeted therapy does not work." Nature Reviews Cancer 12.7 (2012): 487-493.
- McGranahan, Nicholas, and Charles Swanton. "Biological and therapeutic impact of intratumor heterogeneity in cancer evolution." Cancer cell 27.1 (2015): 15-26.
- Zhao, Boyang, et al. "Exploiting temporal collateral sensitivity in tumor clonal evolution." Cell 165.1 (2016): 234-246.
- Imamovic, Lejla, and Morten OA Sommer. "Use of collateral sensitivity networks to design drug cycling protocols that avoid resistance development." Science translational medicine 5.204 (2013): 204ra132-204ra132.
- Nichol, Daniel, et al. "Antibiotic collateral sensitivity is contingent on the repeatability of evolution." Nature communications 10.1 (2019): 1-10.
- Kirkman, Laura A., et al. "Antimalarial proteasome inhibitor reveals collateral sensitivity from intersubunit interactions and fitness cost of resistance." Proceedings of the National Academy of Sciences 115.29 (2018): E6863-E6870.
- Nichol, Daniel, et al. "Steering evolution with sequential therapy to prevent the emergence of bacterial antibiotic resistance." PLoS computational biology 11.9 (2015).
- Hyo-eun, C. Bhang, et al. "Studying clonal dynamics in response to cancer therapy using high-complexity barcoding." Nature medicine 21.5 (2015): 440.
- Gatenby, Robert A., et al. "Adaptive therapy." Cancer Research 69.11 (2009): 4894-4903.
- Kaznatcheev, Artem, et al. "Fibroblasts and alectinib switch the evolutionary games played by non-small cell lung cancer." Nature ecology & evolution 3.3 (2019): 450-456.
© 2026 - The Mathematical Oncology Blog