A global method for molecular dynamics in ABMs
Behind the Paper
A global method for fast simulations of molecular dynamics in multiscale agent-based models of biological tissues
Daniel Bergman, Randy F. Sweis, Alexander T. Pearson, Fereshteh Nazari, Trachette L. Jackson
Read the paper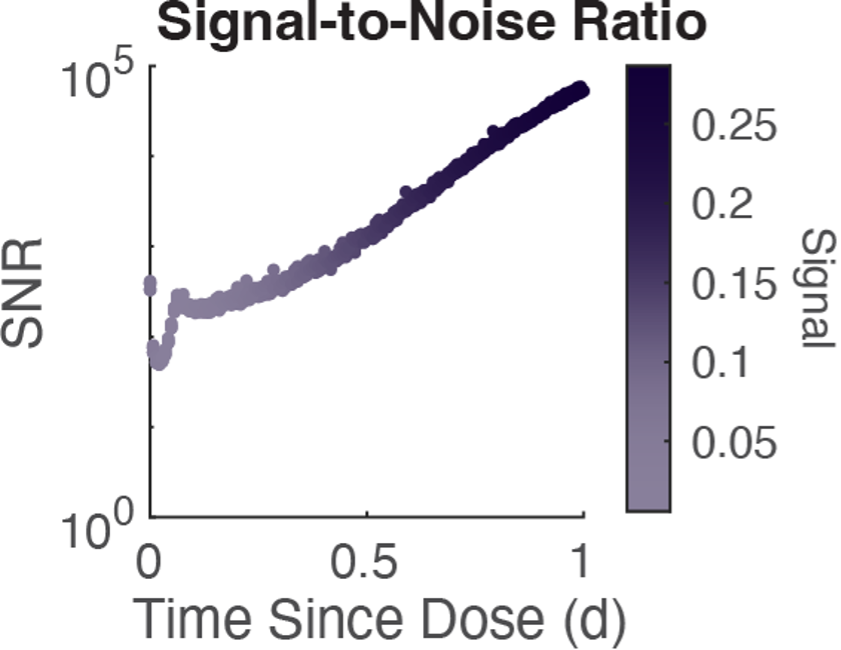
Figure 1: Signal-to-noise ratio is very high, indicating that variability between cells in quite insignificant.
When we looked at the variability in the output of this differential equation across all our agents, we observed that the agents were deviating insignificantly from the mean behavior. That is, solving the equation just once gave a very good approximation for how all cells would respond. Thus, we decided to simply solve this equation just once using the average molecular state variables and then apply that output to all agents. And it worked! We not only observed high levels of agreement between the original method and our new method—dubbed the local and global methods—for solving the molecular dynamics in our model, but the global method was much, much faster.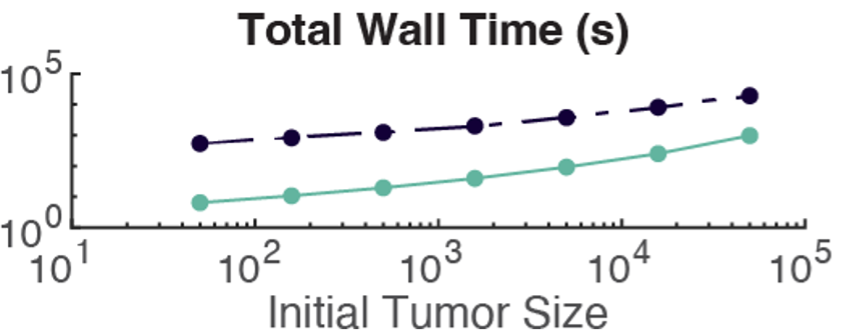
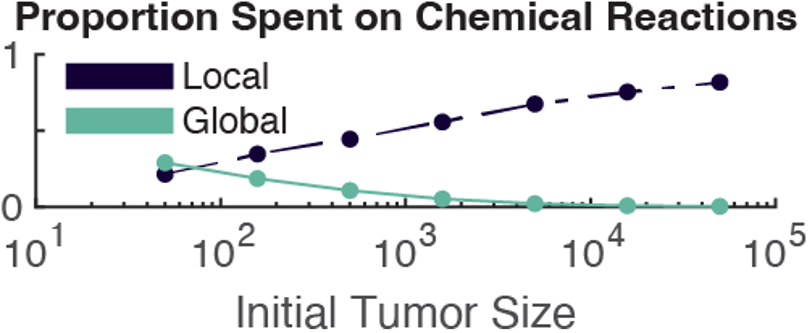
Figure 2: The global method is faster by 1-2 orders of magnitude because it takes less time to solve the chemical reactions. Left) The total wall time for both methods as it depends on the initial number of cells. Right) The proportion of total simulation time spent solving the chemical reactions, not including diffusion.
Now, to be sure, this was only our first effort to “globalize” the solving of these molecular dynamics. But our success propelled us to keep exploring. Since we only ended up using the average concentration of unbound substrate near tumor cells, we partitioned the microenvironment into occupied and unoccupied regions—we refer to these as the tumor-occupied volume (TOV) and the ambient space (AMB) in our paper. Using a standard stencil for solving a 3D partial differential equation (PDE), we simplified the PDE solver down to an ODE solver and still saw strong agreement and even more speedup, though this was not as significant as averaging the cell-by-cell reactions.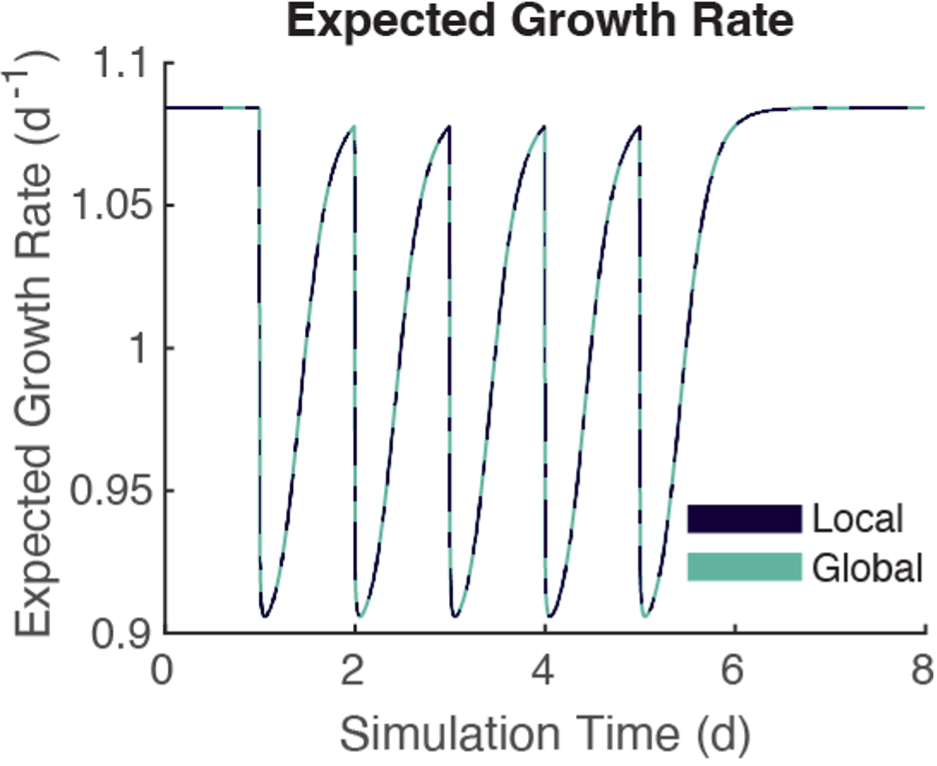
Figure 3: The two methods produce identical growth rates for the agents.
We continued to innovate by developing other ways to improve upon the method. The lowest hanging fruit was partitioning the microenvironment into regions and averaging across all agents within that region. The regions can be defined in any manner and would be best chosen, we believe, to conform to some a priori knowledge of the geometry of the tumor, such as proximity to vasculature. With encouragement from several reviewers, we added the IL-6 model to our paper to show the efficacy in this setting, and even extended our results to look at spatial arrangements of three different phenotypes present in that model.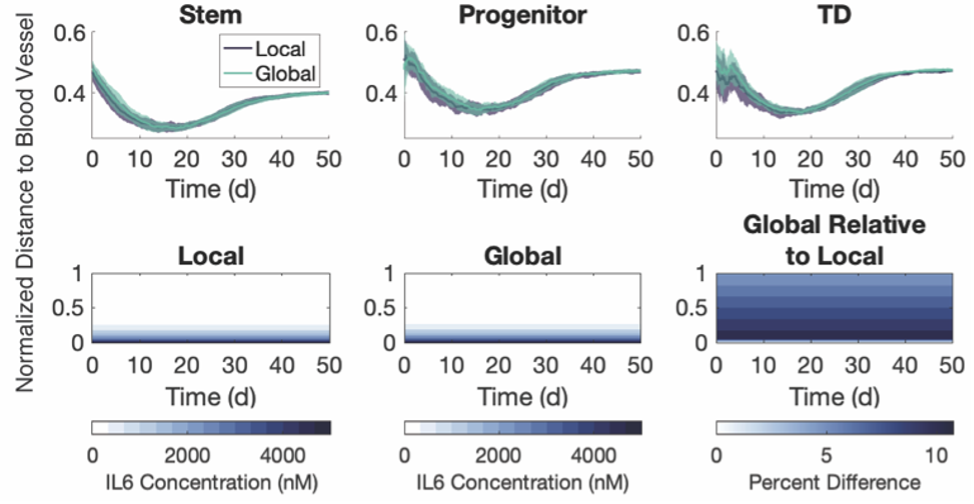
Figure 4: The three phenotypes and the diffusing substrate in the IL-6 model have the same spatial distributions in the two methods. Top row: Average normalized distance to the blood vessel for each simulation. Shaded area indicates +/- 1 SD across 20 simulations. Bottom row: IL-6 concentration averaged across the regions used in the global method throughout one simulation.
The fun doesn’t stop there though! One use case we are currently exploring is how the method works in the context of phenotype switching when molecular concentrations affect cells in a discrete manner. The global method, as presented thus far, seems hard-pressed to achieve the expected phenotypic variability present throughout tumors. However, our upcoming work is focused on showing how the global method works in this context as-is, and works quite well. As a preview, can you tell which of the following was done with the global method?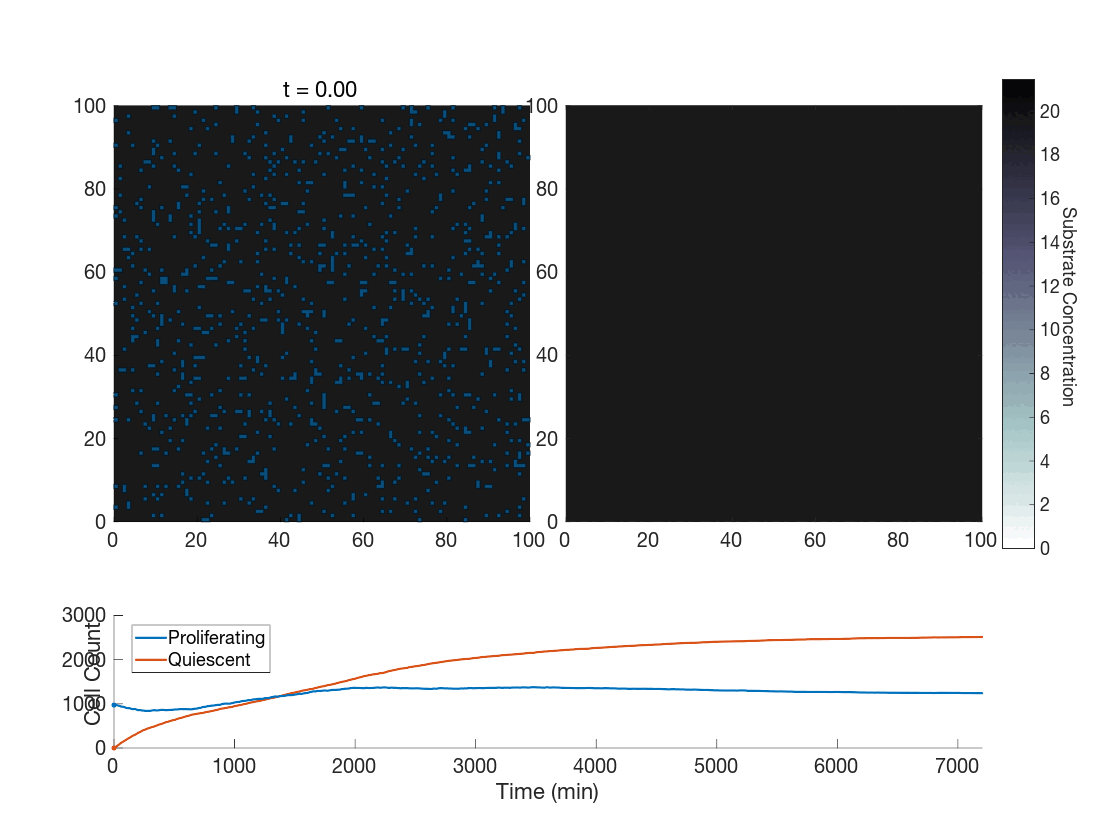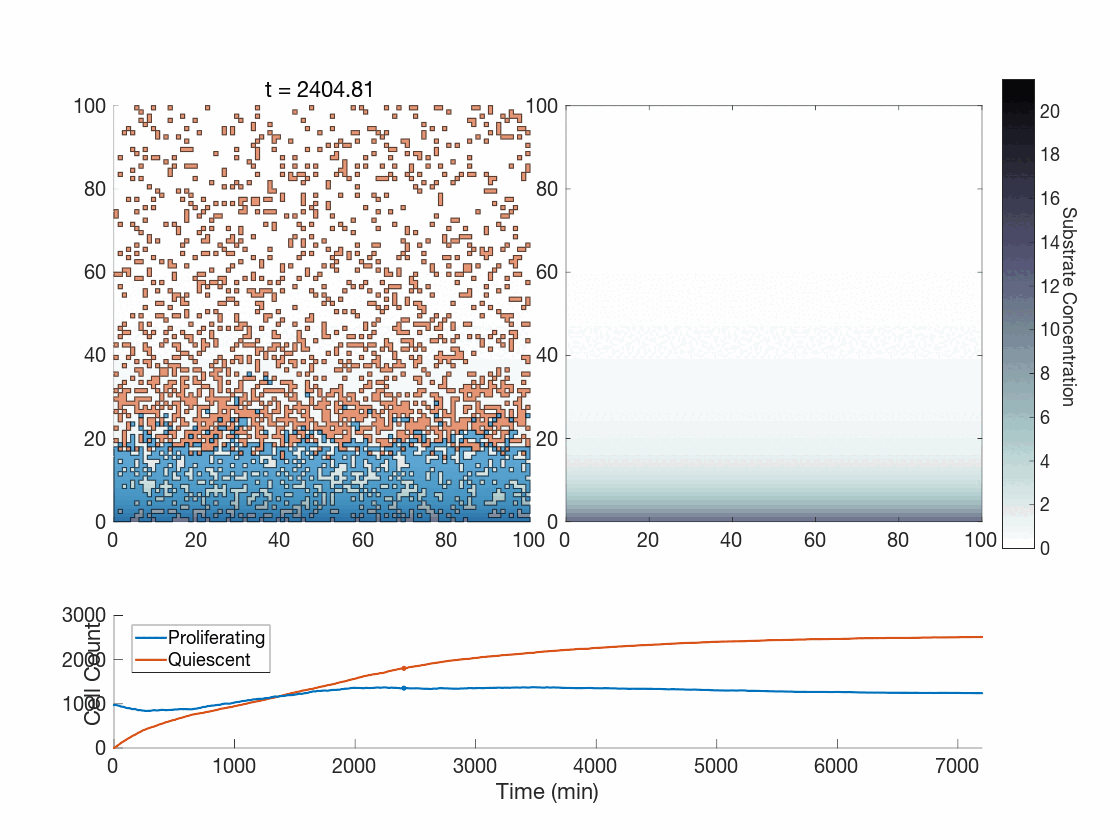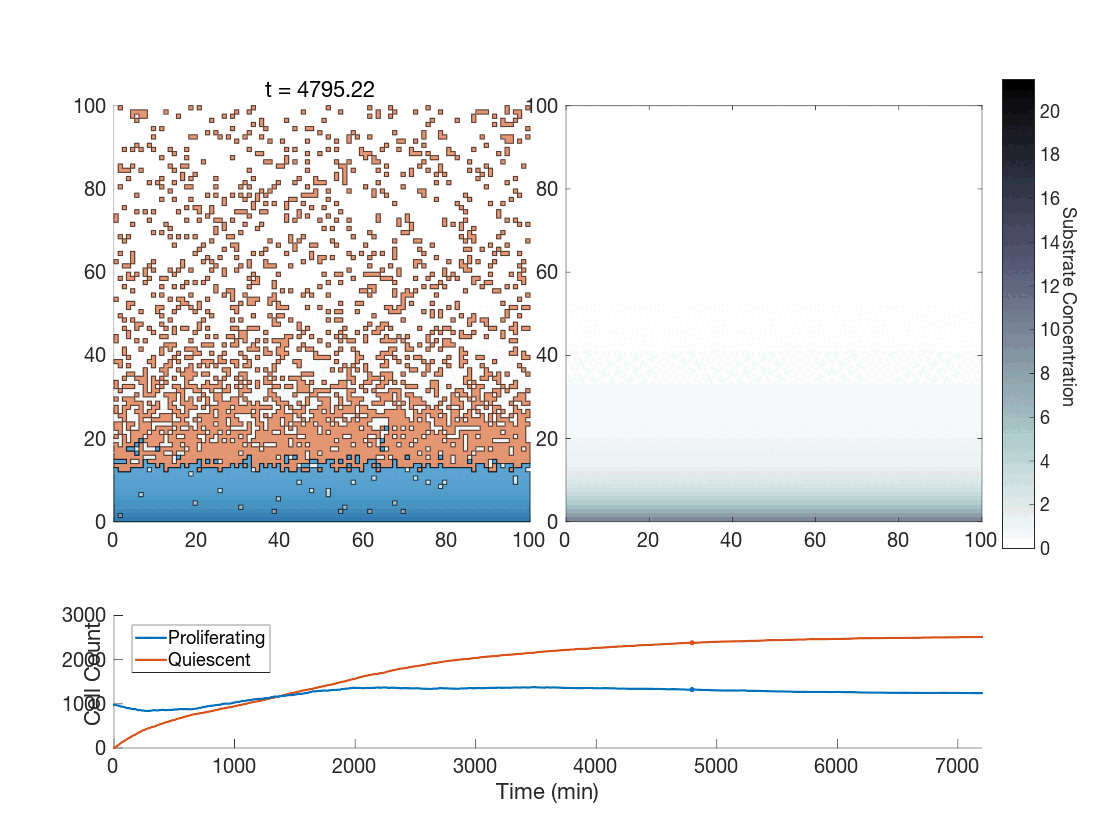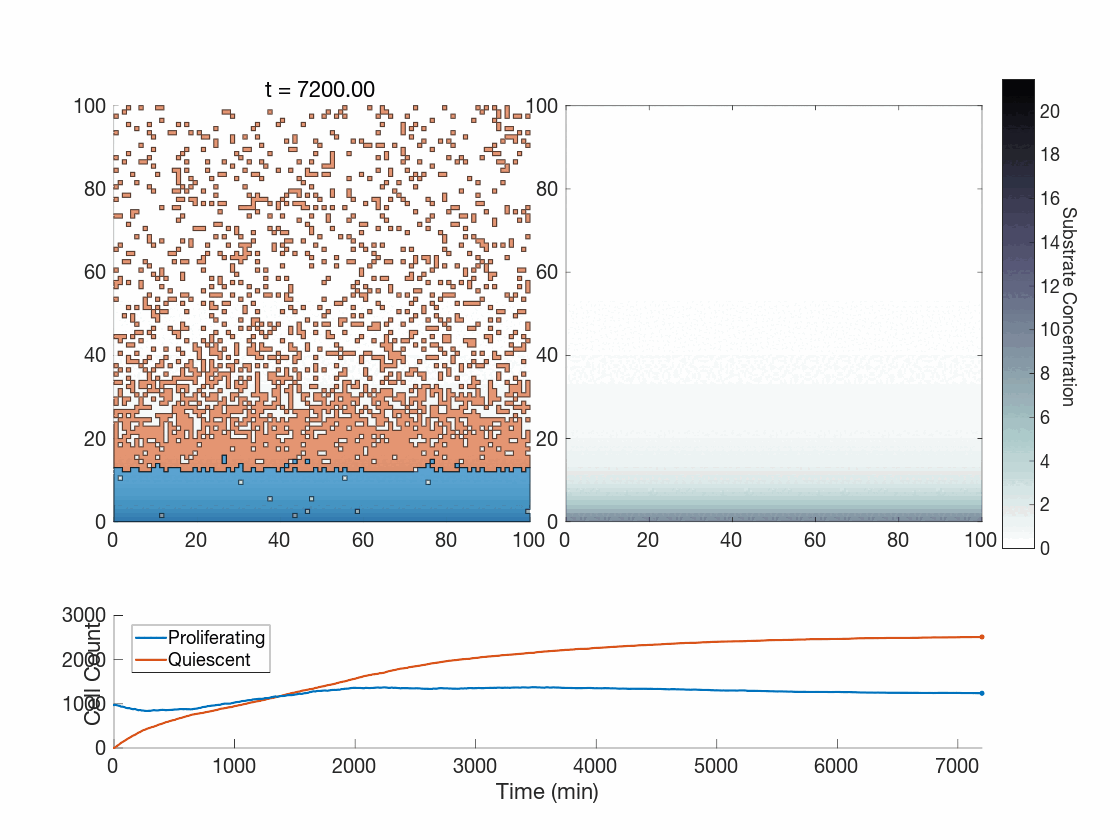
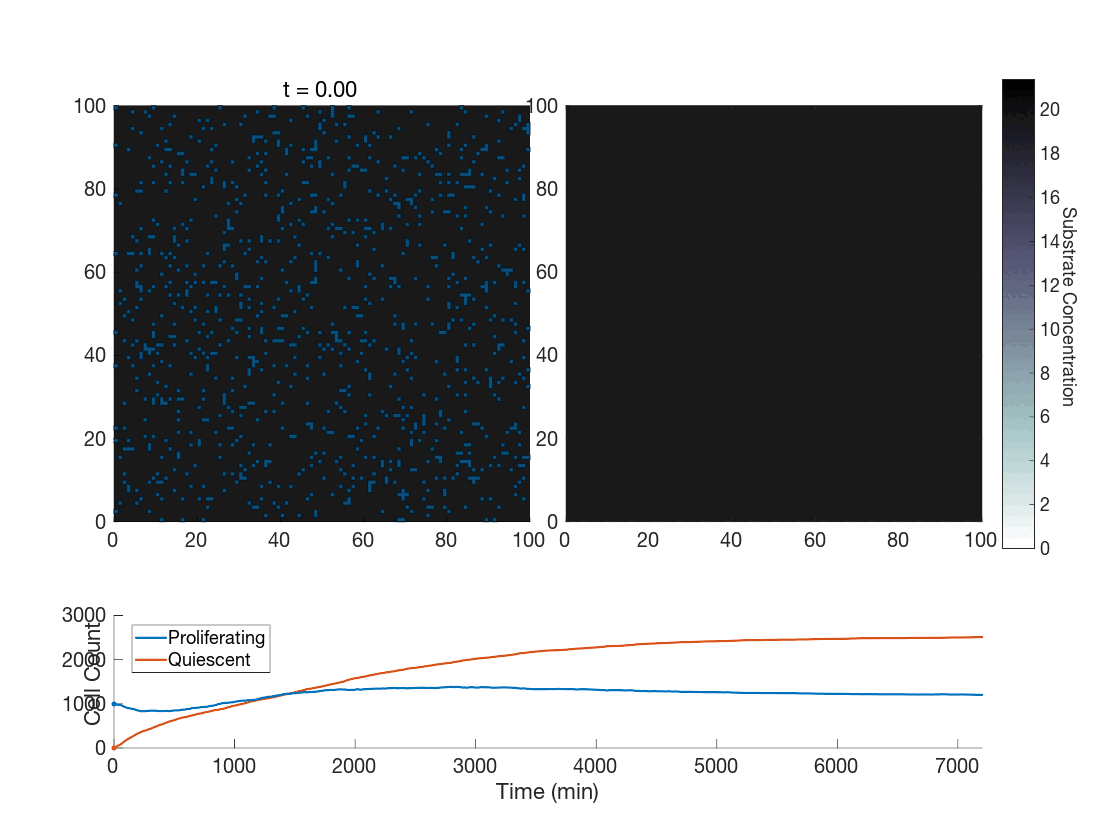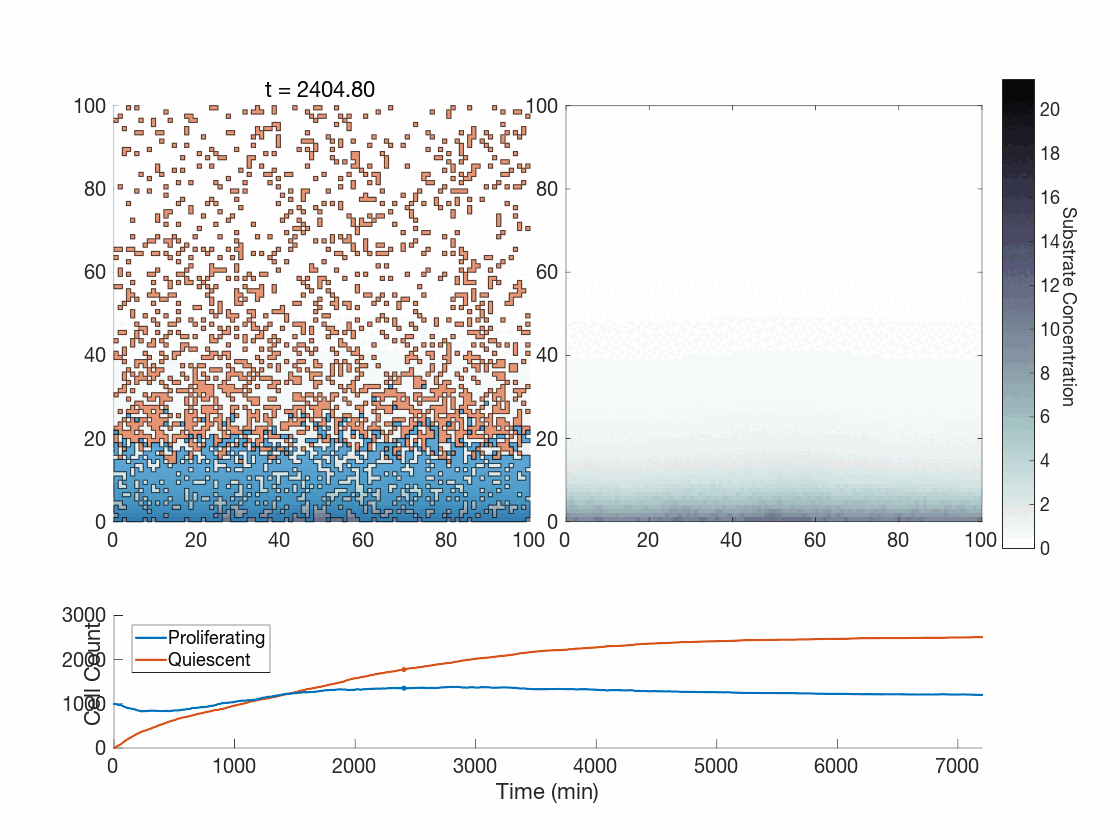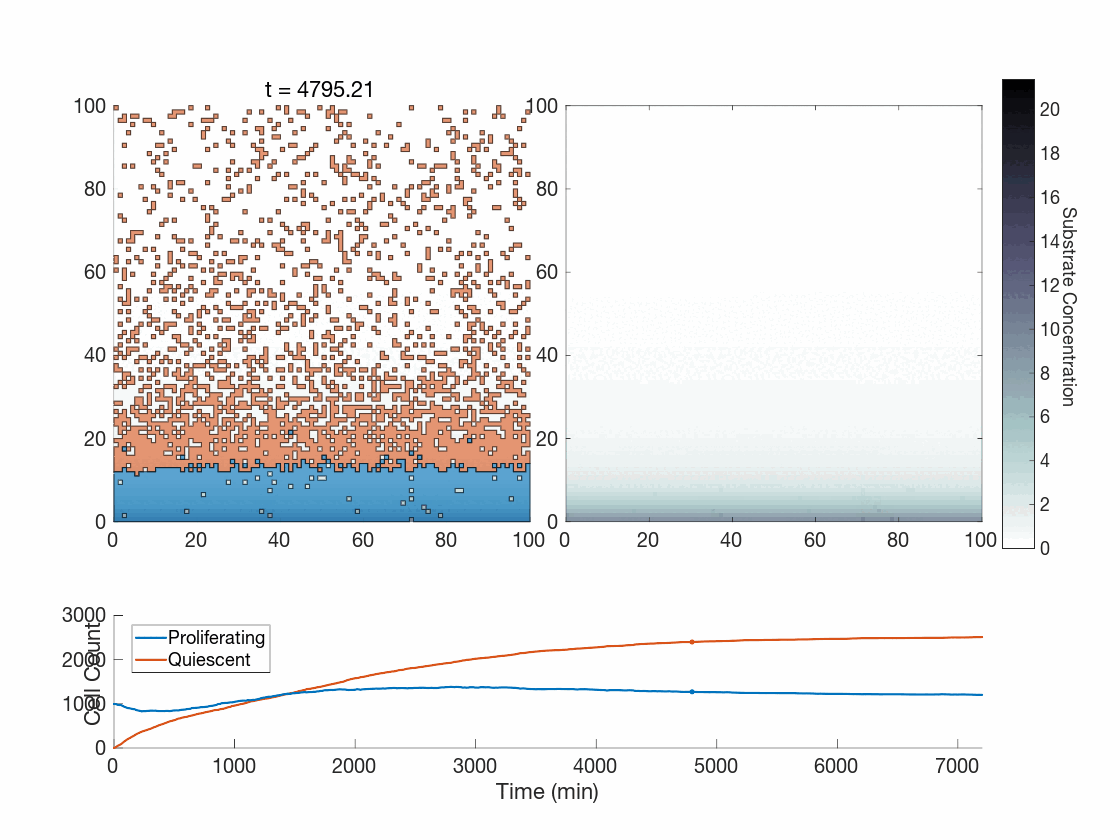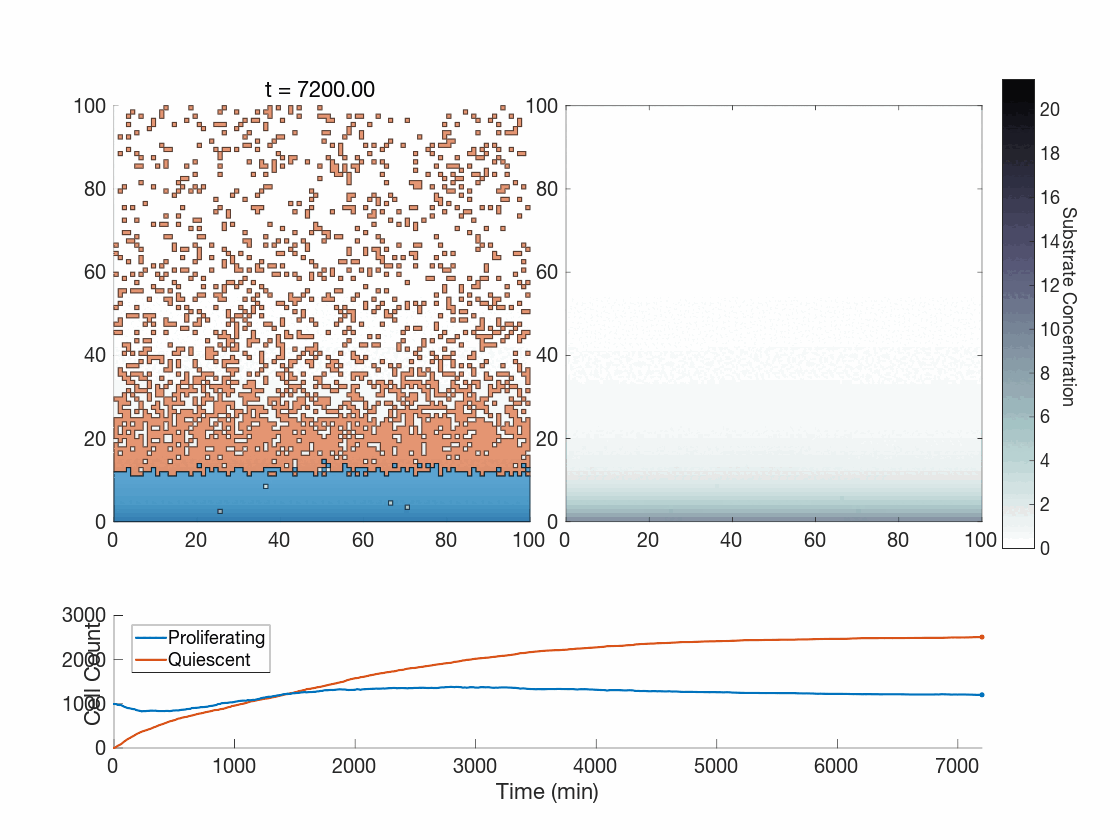
Figure 5: Two simulations of an ABM cells undergoing phenotype switching in response to a diffusing substrate. One uses the global method, but which one? (It seems to be an odd quirk of our converter to .gif that messes up the y-axis label; the .svg files all look fine)
By no means are we done learning how to apply this method to all sorts of ABMs and modeled phenomena. Reactions that involve multiple cells, such as ligand-receptor reactions, pose an intriguing challenge we’re in the process of tackling. Diffusion coefficients are often sensitive to varying microenvironment factors such as the extracellular matrix and would force us to rethink how we define regions and then link them. We also may one day leave the cozy confines of our lattice-based models for the world of off-lattice models, though they too rely on an underlying lattice or mesh which would be a great place to start. We could also start with whatever ABM you’re interested in!© 2026 - The Mathematical Oncology Blog