BIRS Recap: INSITE
In Silico Trials using Simulation and Artificial Intelligence for Therapy Evaluation

This blog is one in a series resulting from the Winter 2025 BIRS workshop on Mechanistic Learning as a Combination of Machine Learning and Modeling in Mathematical Oncology.
Introduction
Clinical trials evaluate the effects of a planned intervention on health-related outcomes. Unfortunately, they are time-consuming, expensive, and often end in failure. However, these failures do not necessarily mean that the therapeutic intervention is ineffective. Rather, it may be due to the selection of a non-optimal treatment schedule or less than ideal patient selection. In silico (or virtual) clinical trials are powerful tools which can address these issues. Specifically, they can be used to: 1. stratify patients according to response (thereby identifying the ideal patient subgroup for a specific therapeutic intervention); 2. investigate and identify the optimal schedule for individual patients, or entire subgroups; and 3. identify beneficial combination therapies.
The routine collection of high-resolution clinical data for cancer patients, from diagnostic imaging to molecular biomarkers and tumor response, creates a unique opportunity for synergy between mechanistic mathematical models and machine learning (ML) in the context of in silico clinical trials. Here, we highlight where such synergy may occur in the in silico trial pipeline (Figure 1) and discuss potential challenges.
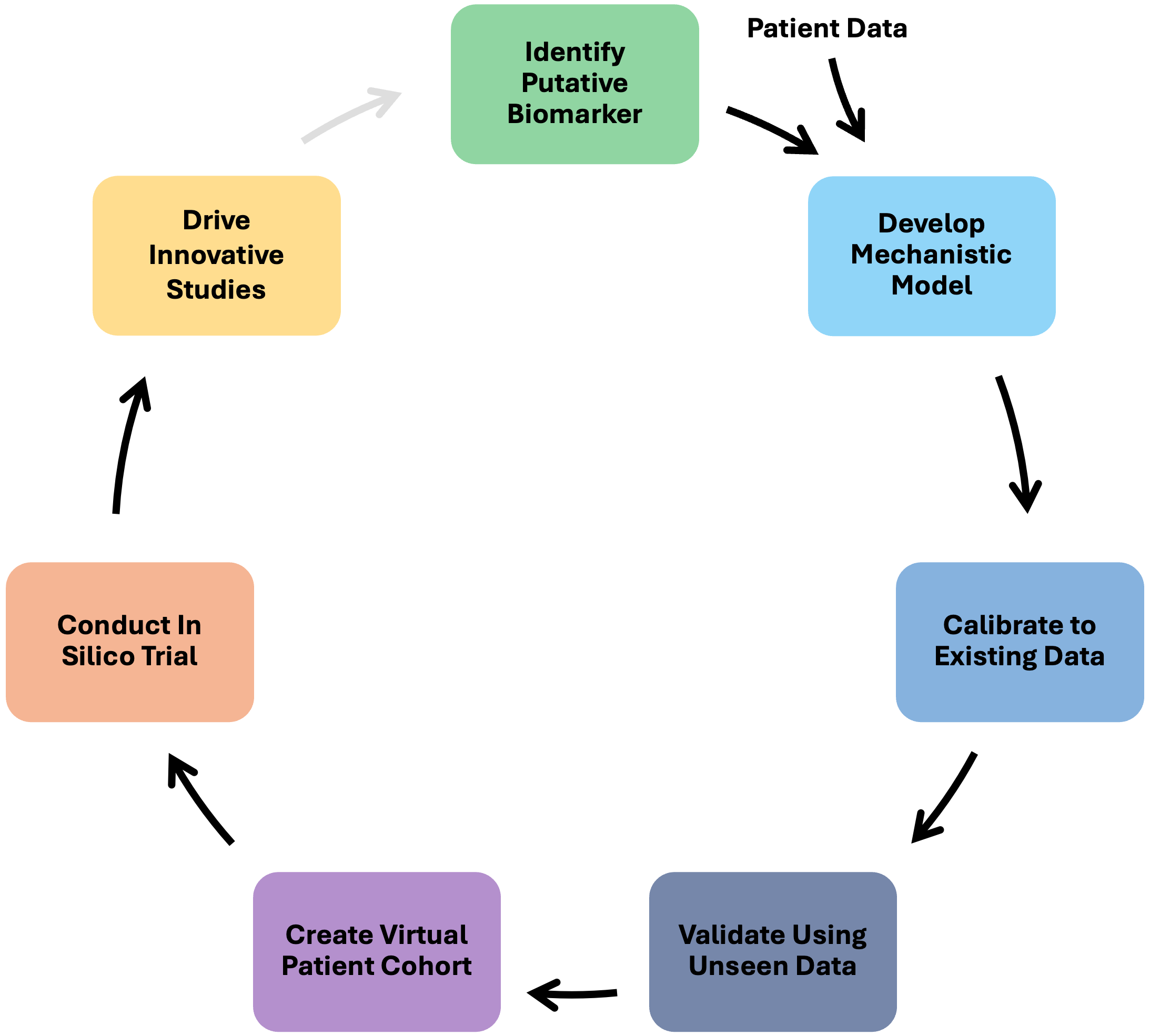
Biomarker Identification
The first step in the in silico clinical trial pipeline is to identify what we’re modeling. That is, we must identify a time resolved biomarker of tumor burden. Ideally, we would directly model tumor burden, but indirect biomarkers (for instance, the molecular marker CA-125 in ovarian cancers) are often easier to obtain, and thus more frequently collected than tumor measurements. Traditionally, the relationships between such data and tumor burden are manually determined and examined. This leads to the identification of a surrogate measure of tumor burden. However, as the breadth, depth and sheer volume of data for each patient increases, this type of manual investigation may soon be impractical. Fortunately, there are multiple ML approaches that can be used in this setting, including recurrent neural networks (RNN), and dynamic time warping (DTW).
RNNs, which were designed for processing sequential data, are well-suited to analyzing and identifying patterns between series of longitudinal data like tumor-progression and levels of molecular markers over time. Alternatively, DTW, which measures similarities between two longitudinal sequences, can be used to investigate relationships between two variables that are not necessarily time aligned. Thus, DTW could be used to identify biomarkers that are early predictors of tumor response, allowing clinicians to intervene prior to tumor progression.
Model Development
The next step in our pipeline is to create a reliable mathematical model that translates biological details – like known mechanisms and available data – into clinically relevant predictions at the individual patient level. This model will serve as the foundation of the in silico clinical trial.
Traditionally, creating these models has been a painstaking manual process, relying heavily on expert knowledge, intuition, and sometimes guesswork. This is where ML can help, as ML methods can accelerate the development of predictive models, making them faster, more objective, and more reliable. However, despite these exciting possibilities, significant challenges remain. ML typically requires extensive, high-quality datasets, which are scarce in biology. Additionally, purely "black-box" approaches, such as support vector machines or neural networks, lack interpretability and can be computationally expensive.
Emerging ML techniques, such as symbolic regression (e.g. AI Feynman), equation learning (e.g., SINDy), and Universal Physics- or Biologically-Informed Neural Networks (UPINNs/BINNs), have the potential to overcome these challenges. They systematically sift through extensive libraries of potential mathematical equations, automatically identifying the simplest and most biologically meaningful candidates. Moreover, these techniques readily incorporate biological constraints, such as conservation laws or physiological boundaries, ensuring interpretability of the results.
Looking ahead, creating standardized libraries of biologically meaningful equations could further streamline model construction, making the process faster and more trustworthy. Equally essential are robust methods for integrating diverse datasets (genomic, cellular, clinical) into unified models and selecting optimal model structures based on both biological and statistical criteria.
Model Calibration
To make accurate predictions about real world phenomena, we need to identify which model inputs or parameters best match available data. In the realm of mathematical oncology, there is a careful balance between finding parameter values that are both biologically plausible and accurately recapitulate the data. While one can manually adjust parameter values to determine combinations that get as close to the data as possible, this process is often time-consuming and inefficient. Minimizing the sum of squared differences (i.e. linear or nonlinear regression) or maximizing the likelihood can also be effective but are often susceptible to overfitting, can be computationally expensive, and do not guarantee convergence to a global minimum.
Several ML techniques have been proposed to address the limitations of traditional methods, leading to automation and greater efficiency. Supervised learning via neural networks, Gaussian processes, random forests, etc. can be used to learn relationships between model parameters and outputs. Such supervised methods essentially learn from examples with the correct behavior (i.e., data). While these are effective, they can be susceptible to overfitting and are dependent on the characteristics of the available data (monotonicity, describing specific treatment conditions, etc). Reinforcement learning can also be used to understand these relationships, but it is accomplished through a trial-and-error approach, receiving rewards or penalties depending on the decisions made. Since there is no predefined “correct answer,” it is less dependent on the available data. Bayesian calibration methods are less focused on fitting the data precisely, but more so on reducing the uncertainty in the model parameters. They can be particularly useful when the model parameters are highly sensitive, but uncertain and when the available data is limited, as is typical in biology.
Model Validation
The model validation step involves assessing how well a model generalizes beyond its training conditions. It is not simply a statistical exercise, but a test of whether the model captures something real about the underlying biology, or merely patterns in a particular dataset.
The traditional approach to validation, which involves partitioning data into training and testing sets or applying the model to an independent cohort, remains necessary but is insufficient. Superficially good performance metrics, like low error rates or high accuracy, may conceal deeper problems. If the external data share structural biases with the training data, we risk providing a false reassurance because of an insufficiently thorough validation process.
Emerging ML methods offer additional tools but do not eliminate the risks. Fairness-aware ML approaches can uncover hidden performance gaps across subpopulations, an important but often overlooked issue in cancer modeling. Feature similarity metrics, drawn from ML, can quantify whether model-generated outputs truly resemble biological data, offering a more sensitive measure than global error rates alone. When used with care, these tools could improve confidence in the validation provided. However, an inexpert application of these tools can potentially add complexity without adding insight.
The main challenge inherent in the validation step is the lack of consensus as to what constitutes sufficient validation. In addition, access to truly independent, high-quality validation data is limited, and in some fields, perhaps prohibitively so. Finally, there is a temptation to over-automate validation workflows and to rely on ML-driven pipelines that bypass human scrutiny. An over-reliance on automation could turn validation into an opaque process that may not catch biologically relevant issues.
Validation should not be treated as a final hurdle before model publication or deployment. Instead, we should treat the validation step as a continuous, iterative process that adapts as models are applied in new settings. Our goal should not be merely to achieve technical success, but to build confidence that the model represents the phenomena it aims to predict.
Virtual Population Creation
Once the mathematical model has been preliminarily validated, it’s time to construct the virtual population. A virtual population can be thought of as a collection of “plausible patients”, defined as parameterizations of the mathematical model that produce outputs consistent with known biological behavior. For a cancer model, this would minimally be constraints on the tumor biomarker time series, though the precise constraints imposed depend on the data available.
There are challenges that arise at this step of an in silico clinical trial that ML can help address. A sufficiently large pool of virtual patients is created by generating many random parameterizations of the model, and testing the parameterizations for feasibility. For more complex mathematical models, the associated computational demands can become prohibitively expensive. A surrogate ML model that approximates the output of a mechanistic model using a less computationally intensive framework (like a neural network) can be quite helpful here. Such a surrogate model that is trained on the full mathematical model can be used to rapidly pre-screen for parameterizations that result in plausible patients.
The use of surrogate models to facilitate the creation of plausible patients is not without its own challenges. Training a surrogate model is computationally intensive in its own right, so one needs to consider when those computational costs are more prohibitive than a direct approach to generating plausible patients. Further, any error in the surrogate model’s representation of the true model could skew the composition of the generated virtual population and the results of your in silico clinical trial.
Conducting the In Silico Clinical Trial and Learning from the Results
At this point, the in silico clinical trial can be conducted by solving the validated mathematical model for each virtual patient. This process generates a lot of data to be analyzed. This is a very natural place to use ML, as ML techniques are designed to learn patterns from high dimensional data. For instance, we may want to try and determine what features (parameters) of a virtual patient help stratify patients into different groups, such as complete responders, partial responders, and non-responders. We may also want to uncover rare subpopulations, like those who respond exceptionally well or poorly to treatment.
Logistic regression is a simple and interpretable ML tool that can be used to determine which features of a virtual patient are most important in deciding its classification into a specific subgroup. If one is interested in learning more complex relations between parameters, and how they influence patient classification, sets of decision trees (referred to as random forests) can help identify combinations of features that best explain the stratification of the virtual patients. Another tool to consider is SHAP (SHapley Additive exPlanations), which assigns each feature a numerical value representing its contribution to the outcome of interest. It can also be used to understand how these features interact to influence the classification of a virtual patient. While these and other ML tools can help uncover complex patterns, it is important to remember that the reliability of these predictions is inherently constrained by the quality of the data generated from the in silico clinical trial.
Conclusion
In summary, there are multiple opportunities for synergy between machine learning and mathematical modeling in the context of in silico clinical trials. However, there are also significant challenges. ML approaches are often highly sensitive to noise, and generally require large amounts of high quality data, which can be difficult to acquire in clinical settings. Additionally, many ML techniques operate as black-boxes, which lack interpretability and generalizability, making it challenging to understand and generalize the results and predictions in biological settings. Furthermore, these approaches generally have high computational costs. If the time it takes to train and validate the ML model exceeds the time it would take to complete that step in the pipeline without ML, then integrating ML into the pipeline at that step may be impractical.
Despite these challenges, it is clear that the thoughtful use of ML tools can result in richer and more informative in silico clinical trials, potentially speeding up drug development, improving reliability, and helping patients get effective treatments faster. This is an exciting and critical time to explore how to judiciously introduce ML into the in silico trial pipeline. At the same, we underscore that ML should not replace the expertise of the modeller. Finding the right balance is key, as the potential payoff of integrating machine learning and modeling is too important to miss as in silico clinical trials play an increasingly important role in the drug development pipeline.
Important Papers
- Benzekry, S. (2020). Artificial intelligence and mechanistic modeling for clinical decision making in oncology. Clinical Pharmacology & Therapeutics, 108(3), 471–486. doi: 10.1002/cpt.1951
- Brady, R., & Enderling,H. (2019). Mathematical models of cancer: When to predict novel therapies, and when not to. Bulletin of Mathematical Biology, 81(10), 3722–3731. doi: 10.1007/s11538-019-00640-x
- Metzcar, J., Jutzeler, C. R., Macklin, P., Köhn-Luque, A., & Brüningk, S. C. (2024). A review of mechanistic learning in mathematical oncology. Frontiers in Immunology, 15, 1363144. doi: 10.3389/fimmu.2024.1363144
- Morgan, C., Gevertz, J. L., Kareva, I., & Wilkie, K. P. (2023). A practical guide for the generation of model-based virtual clinical trials. Frontiers in Systems Biology, 3. doi: 10.3389/fsysb.2023.1174647
- Norton, K.-A., Bergman, D., Jain, H. V., & Jackson, T. (2025). Advances in surrogate modeling for biological agent-based simulations: Trends, challenges, and future prospects. arxiv. doi: 10.48550/arXiv.2504.11617
© 2026 - The Mathematical Oncology Blog